Marcela Godoy1 ID· Carola López2 ID· Felipe Álvarez Chavez3 ID· Raquel Borges Pinto4 ID Verónica Botero Osorio5 ID· María Valentina Dolz Aguilar6 ID· Michelle Higuera7 ID· Reynaldo de Jesús Michel Aceves8 ID· Gloria Ríos Marcuello9 ID· Lorena Rodríguez González10 ID Claudia Rojo Lillo11 ID· Humberto E Soriano12 ID· Mirta Ciocca13 ID
1 Hospital Clínico San Borja Arriaran. Santiago de Chile. Chile.
2 Centro Hospitalario Pereira Rossell. Montevideo. Uruguay.
3 Hospital de Pediatría, Centro Médico Nacional de Occidente, IMSS. Guadalajara, Jalisco. México.
4 Hospital da Criança Conceição do Grupo Hospitalar Conceição. Porto Alegre, Rio Grande do Sul. Brasil.
5 Unidad de Trasplantes Fundación Valle de Lili. Cali. Colombia.
6 Hospital de Carabineros y Clínica Las Condes. Santiago de Chile. Chile.
7 Hospital San José Infantil. Bogotá. Colombia.
8 Hospital Central Militar. Ciudad de México. México.
9 Clínica Alemana de Santiago, Facultad de Medicina Clínica Alemana. Universidad del Desarrollo. Santiago de Chile. Chile.
10 Hospital San Juan de Dios y Clínica Alemana de Santiago. Santiago de Chile. Chile.
11 Hospital Regional de Antofagasta y Universidad de Antofagasta. Antofagasta. Chile.
12 Departamento de Gastroenterología y Nutrición Pediátrica. Pontificia Universidad Católica de Chile. Santiago de Chile. Chile.
13 Hospital Alemán. Buenos Aires. Argentina.
Acta Gastroenterol Latinoam 2022;52(3):344-354
Recibido: 26/11/2021 / Aceptado: 08/08/2022 / Publicado online el 29/09/2022 / https://doi.org/10.52787/agl.v52i3.134

Resumen
La ictericia neonatal requiere el diagnóstico diferencial entre la hiperbilirrubinemia no conjugada o indirecta y la hiperbilirrubinemia directa o conjugada, característica de la colestasis neonatal, que siempre es patológica. Se recomienda en todo recién nacido de 15 días o más de vida con ictericia realizar bilirrubina diferencial para poder detectarla. La colestasis se define por bilirrubina directa > 1 mg/dl (17 µmol/L) y su frecuencia estimada durante el período neonatal es de 1:2.500 recién nacidos vivos. Las etiologías más frecuentes son: infecciosas, genéticas/metabólicas, obstructivas, endocrinológicas, tóxicas, inmunológicas. La causa más común es la atresia biliar, que se manifiesta clínicamente por ictericia y acolia, por lo que frente a estos signos siempre debe plantearse este diagnóstico como primera opción a descartar.
Es fundamental que el diagnóstico de colestasis neonatal sea precoz para optimizar el manejo en las causas tratables y mejorar su pronóstico. Desafortunadamente, no siempre el diagnóstico es precoz. Es por esto que el Grupo de Trabajo de la Sociedad Latinoamericana de Gastroenterología, Hepatología y Nutrición Pediátrica realizó esta revisión de diagnóstico, tratamiento y seguimiento de la colestasis neonatal, cuyo objetivo es proporcionar una herramienta útil que permita el diagnóstico temprano y mejorar el pronóstico de los niños con colestasis neonatal.
Palabras claves. Ictericia, colestasis neonatal, atresia biliar.
Neonatal Cholestasis: A Narrative Review of the Working Group of the Latin American Society of Pediatric Gastroenterology, Hepatology and Nutrition
Summary
Neonatal jaundice requires a differential diagnosis between unconjugated or indirect hyperbilirubinemia and direct or conjugated hyperbilirubinemia, characteristic of neonatal cholestasis, which is always pathological. It is recommended in all newborns of 15 days or older with jaundice to perform differential bilirubin to be able to detect it. Cholestasis is defined by direct bilirubin > 1 mg/dl (17 µmol/L) and its estimated frequency during the neonatal period is 1:2,500 live births. The most frequent etiologies are infectious, genetic/metabolic, obstructive, endocrinological, toxic, immunological. The most common cause is biliary atresia, which is clinically manifested by jaundice and acholia, so in patients with these signs that diagnosis should always be considered as the first option to rule out. An early diagnosis of neonatal cholestasis is essential to optimize management of treatable causes and improve prognosis. Unfortunately, diagnosis is not always early. That is why the Working Group of the Latin American Society of Pediatric Gastroenterology, Hepatology and Nutrition carried out this review of diagnosis, treatment and follow-up of neonatal cholestasis, whose objective is to provide a useful tool that allows early diagnosis and improves the prognosis of children with neonatal cholestasis.
Keywords. Jaundice, neonatal cholestasis, biliary atresia.
Abreviaturas
CN: Colestasis neonatal.
AB: Atresia biliar.
GGT: Gamma glutamil transpeptidasa.
TH: Trasplante hepático.
CIFP: Colestasis intrahepática familiar progresiva.
Introducción
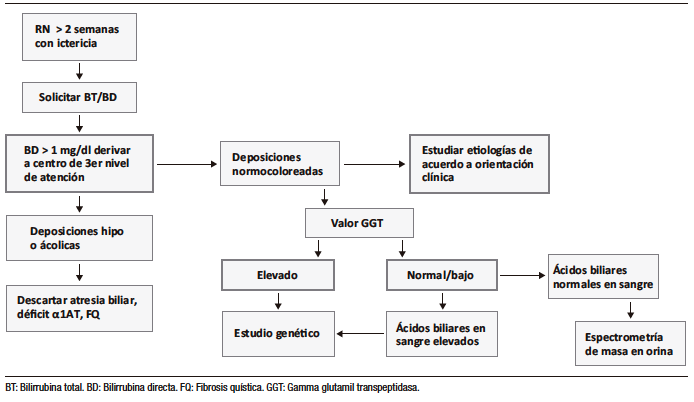
La colestasis neonatal (CN) ocurre como consecuencia de una disminución del flujo y/o excreción biliar, con la consiguiente acumulación de sustancias que normalmente son excretadas por la bilis hacia el intestino. Desde el punto de vista bioquímico, se caracteriza por un nivel de bilirrubina directa en sangre > 1 mg/dl (17 µmol/L).
La CN es un trastorno poco frecuente, con una incidencia estimada de 1:2.500 recién nacidos vivos, siendo mayor en prematuros.
La presencia de ictericia a partir de las 2 semanas de vida obliga a realizar el fraccionamiento de la bilirrubina para diagnosticar si la ictericia es de causa fisiológica o por leche materna (aumento de bilirrubina indirecta) o si corresponde a colestasis.
La ictericia colestática siempre es patológica; de ahí la importancia de realizar un diagnóstico rápido y comenzar precozmente con el tratamiento específico en aquellas etiologías que lo requieran. Del diagnóstico oportuno depende en muchos casos la sobrevida del niño.1-3
Este artículo es el resultado de la revisión realizada por el Grupo de Trabajo de la Sociedad Latinoamericana de Gastroenterología, Hepatología y Nutrición Pediátrica. Se destacan los avances alcanzados en los últimos años en un tema muy complejo.
Etiologías de colestasis neonatal
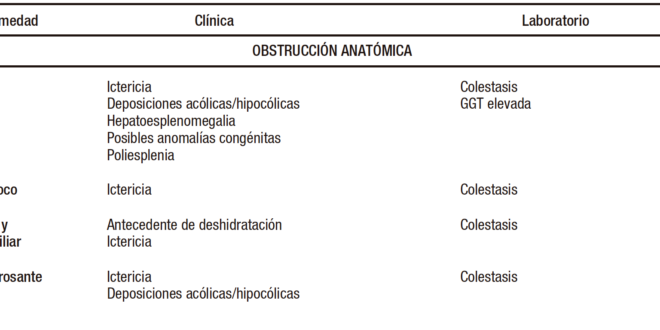
Las enfermedades que causan CN son un grupo muy numeroso y diverso de entidades infecciosas, genético-metabólicas, obstructivas, endocrinológicas, tóxicas (alimentación parenteral, fármacos) e inmunológicas (Tabla 1).
Tabla 1. Diagnóstico diferencial de colestasis neonatal
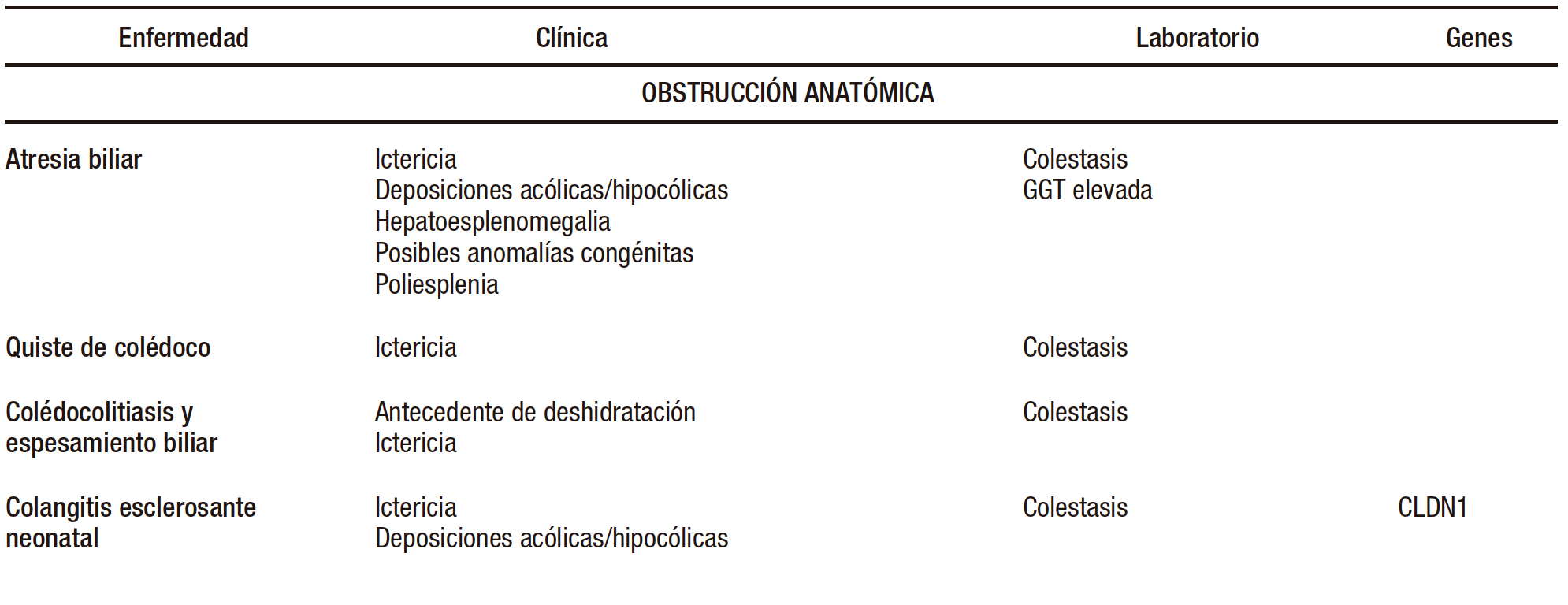
Al identificar a un recién nacido o lactante pequeño con colestasis, los primeros estudios estarán dirigidos a descartar las causas más frecuentes y tratables. Las causas más comunes son la atresia biliar (AB) 25-45% y una variedad creciente de enfermedades genéticas.2-4
Las causas más importantes de CN serán revisadas brevemente.
1) Obstrucción anatómica
1.1 Atresia biliar
La AB es la causa tratable más frecuente de CN (25-45%) y es la principal causa de trasplante hepático pediátrico en el mundo. Se estima una prevalencia de entre 1:6000 y 1:19000 recién nacidos vivos, variable según las regiones. Es más común en países asiáticos. Predomina en el sexo femenino.
La AB es el resultado de un proceso fibroinflamatorio progresivo que afecta a los conductos biliares intra y extrahepáticos y culmina en el desarrollo de cirrosis biliar. Ocasiona la muerte antes de los 3 años de vida si no se realiza precozmente la portoenteroanastomosis u operación de Kasai. Se describen las siguientes variantes clínicas de AB: 1) la forma no sindrómica (84%), la más frecuente 2) AB con una malformación, pero sin defectos de lateralidad (6%) y 3) la forma sindrómica con defectos de lateralidad (10%). Los grupos 2 y 3 tienen otras anomalías asociadas, predominantemente cardiovascular (16%) y gastrointestinal (14%).2-6
La etiología de la AB es desconocida y se han propuesto diferentes teorías patogénicas: genética, infección viral, toxinas, injuria inflamatoria crónica o autoinmune de los conductos biliares.
Las características clínicas de la enfermedad son las siguientes:
• Ictericia progresiva
• Acolia
• Coluria
• Hepatomegalia de consistencia aumentada, acompañada en ocasiones de esplenomegalia.
Al nacimiento los niños suelen tener aspecto normal. Presentan ictericia en forma temprana, pero es común que se confunda con ictericia fisiológica o ictericia por leche materna. La diferencia es que en la AB la ictericia persiste y se asocia a deposiciones hipo o acólicas y hepatoesplenomegalia. La afectación del peso depende del tiempo de evolución de la enfermedad.6-8
Los hallazgos de laboratorio, inespecíficos, son los siguientes:
• Bilirrubina total: 6 a 10 mg/dl.
• Bilirrubina directa: 3 a 8 mg/dl.
• Transaminasas moderadamente aumentadas.
• Gamma glutamil transpeptidasa (GGT) elevada.2-4
Los siguientes exámenes complementarios van a contribuir al diagnóstico de la enfermedad:
• Ecografía abdominal: realizada con cuatro horas de ayuno permite definir la forma y tamaño de la vesícula. Su ausencia puede sugerir, pero no confirma AB. El área triangular o signo del cordón fibroso (área ecogénica del porta hepatis) tiene una sensibilidad del 70% al 100% y una especificidad del 98% al 100% para el diagnóstico de AB. La ecografía permite, además, diagnosticar la existencia de poliesplenia, vena porta preduodenal, agenesia de la vena cava, etcétera.2, 6, 7
• Biopsia hepática: tiene gran valor diagnóstico (especificidad del 95%), si se realiza con una muestra apropiada (más de 10 espacios porta) y cuando es informada por un patólogo con experiencia pe-diátrica. La histología se caracteriza por la presencia de proliferación ductular, tapones biliares en los conductos interlobulillares y fibrosis portal. Suele incluirse al realizar la colangiografía intraoperatoria (estándar dorado/gold standard) para no demorar el diagnóstico de certeza de AB.1, 2, 8, 9
• Cuantificación en sangre de matrix metaloproteinasa 7. Esta proteasa se asocia a fibrosis en pacientes con AB. Ha demostrado tener buena precisión para diagnóstico de AB por encima de un valor de corte determinado, con un valor predictivo positivo del 92%. Promete una futura aplicación clínica que, se espera, abreviará el algoritmo diagnóstico.10, 11
La colangiografía intraoperatoria y la visualización histológica del remanente ductal permiten confirmar el diagnóstico. En alrededor del 20% de los casos, dicho estudio puede sugerir un diagnóstico incorrecto. Esto ocurre en pacientes con hipoplasia del tracto biliar, como en el síndrome de Alagille, fibrosis quística o deficiencia de alfa-1-antitripsina, por lo cual es fundamental la experiencia del equipo quirúrgico. Al confirmarse el diagnóstico de AB se procede a la realización de la operación de Kasai.2, 3, 9
La AB es una urgencia quirúrgica. En la actualidad se aconseja un tratamiento secuencial, inicialmente la cirugía de Kasai, que es una portoenteroanastomosis en Y de Roux. La edad en la que se realiza este procedimiento (idealmente antes de los 45 días de vida) en conjunto con la experiencia del equipo quirúrgico son los factores más importantes para la sobrevida del paciente con el hígado nativo. Si el Kasai fracasa es necesario indicar un trasplante hepático (TH). Si la ictericia desaparece a los 3 meses después de la cirugía, las tasas de supervivencia sin TH a los 10 años son del 75% al 90%; si la ictericia persiste, la tasa de supervivencia sin TH a los 3 años es del 20%.2, 3, 9
Dada la frecuencia del diagnóstico tardío, se ha buscado implementar métodos de cribado (screening) para AB que permitan llegar a la cirugía antes de los 45-60 días de vida. La detección del lactante ictérico con deposiciones acólicas, mediante el método de tamizaje con tarjetas colorimétricas de las deposiciones, es simple, no invasivo y de bajo costo. Su implementación en países con alta incidencia ha disminuido el tiempo de referencia y cirugía en estos niños. La detección la realizan los padres, comparando el color de las deposiciones del recién nacido con los diferentes colores de las imágenes incluidas en la cartilla, que son fotografías de heces acólicas e hipocólicas (asociadas a colestasis) y otras características del niño normal. (Figura 1).12-15
Figura 1. Colorimetria de deposiciones

1.2 Otras
Quiste de colédoco, litiasis vesicular, barro biliar, bilis espesa, perforación espontánea del colédoco, colangitis esclerosante neonatal (herencia autosómica recesiva).2, 3, 6
2) Infecciosas
Considerar infecciones bacterianas (infección urinaria, sepsis, sífilis), virales (citomegalovirus, herpes simple, rubeola, adenovirus, parvovirus, enterovirus, etc.) y parasitaria (toxoplasmosis).2, 16, 17
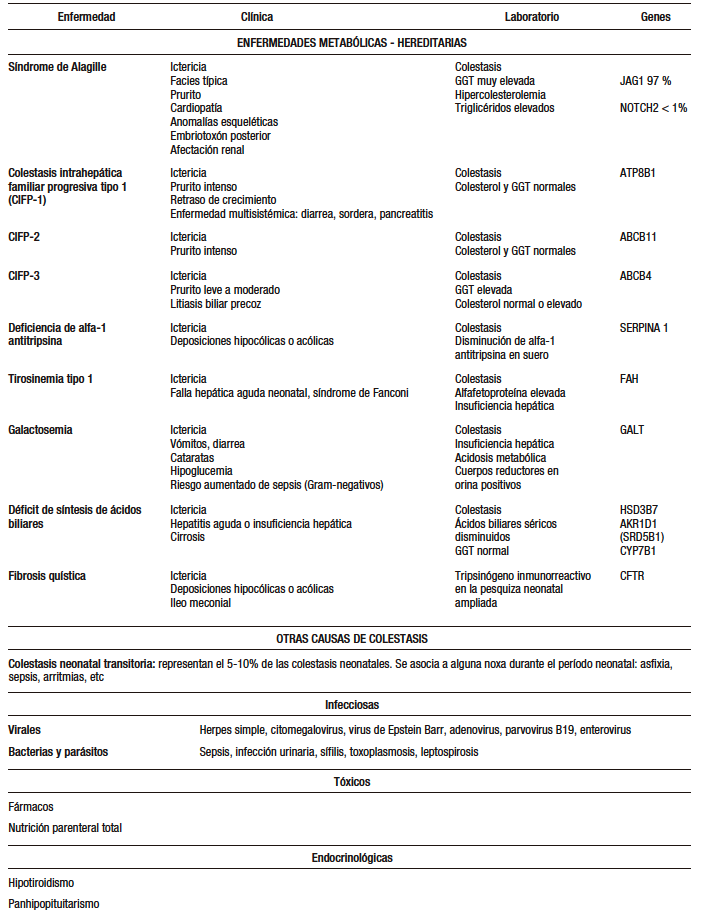
3) Enfermedades metabólicas hereditarias
La última década ha revolucionado el abordaje de los pacientes con CN en relación con los nuevos diagnósticos de causas genéticas. Estos han sido facilitados por los avances en bioquímica y biología molecular.
3.1 Deficiencia de alfa-1-antitripsina
Es la causa genética más frecuente de enfermedad hepática. Es una enfermedad autosómica recesiva, en la cual los dos alelos deben estar mutados para que se manifieste. El diagnóstico se realiza con la determinación del fenotipo (normal: MM; anormal: ZZ o SZ; heterocigota: MZ, MS). La proteína anómala se polimeriza y es retenida en el hepatocito. Los niños con fenotipo ZZ y SZ pueden desarrollar enfermedad hepática y entre el 10-15% se presentan con CN. La colestasis suele ser severa y la presencia de heces acólicas puede plantear el diagnóstico diferencial con AB. La ictericia suele desaparecer espontáneamente en la mayoría de los pacientes alrededor del cuarto mes de vida. El nivel sérico de alfa-1-antitripsina suele estar descendido. El estudio del fenotipo es una modalidad diagnostica confiable. En la biopsia hepática se identifican gránulos PAS (periodic acid-Schiff) positivos diastasarresistentes, en la región periportal, pero pueden no estar presentes antes de los 3 meses de vida. Los pacientes que presentan CN (10-30%) desarrollarán moderada o severa enfermedad hepática durante la infancia y pueden requerir un TH.17-19
3.2 Síndrome de Alagille
El Síndrome de Alagille es una enfermedad multisistémica, autosómica dominante con penetrancia variable y pobre correlación genotipo/fenotipo. Se estima que el 15% son mutaciones de novo. La afectación hepática, caracterizada por el patrón histológico de pobreza de conductos biliares interlobulares (ductopenia), se asocia con otras manifestaciones. Representa el 2 al 14% de las CN. La mayoría de los pacientes se presenta con colestasis, hipercolesterolemia, con o sin heces hipo/acólicas y retraso del crecimiento. El diagnóstico incluye facies características (frente amplia, mentón puntiagudo, hipertelorismo), embriotoxón posterior, vértebras en ala de mariposa, enfermedad renal, cardiopatía (más frecuentemente estenosis pulmonar periférica).
La pobreza de conductos biliares está presente solamente en el 60 % de las biopsias hepáticas en menores de 6 meses y la proliferación de conductos biliares es un hallazgo frecuente en estas.2, 17, 20, 21
3.3 Colestasis intrahepática familiar progresiva
La colestasis intrahepática familiar progresiva (CIFP) es un grupo de enfermedades monogénicas no relacionadas, en las cuales uno de los genes involucrados en el transporte canalicular hepatobiliar ocasiona colestasis progresiva y daño hepático. Este grupo de entidades autosómicas recesivas incluye las CIFP de tipos 1, 2 y 3, las cuales son el resultado de la acumulación de sales biliares dentro de los hepatocitos. Un hallazgo clínico característico de los pacientes con CIFP 1 y 2 (debido a deficiencias de los genes ATP8B1 y ABCB11 respectivamente) es la presencia de GGT normal o disminuida, asociada con colesterol sérico normal o bajo. Son de inicio más temprano y presentan prurito intenso. Los pacientes con CIFP 3 (deficiencia de ABCB4) tienen GGT elevada y grado variable de colestasis, sin prurito.2, 17, 20-24
En los últimos años, se han descrito tres nuevas CIFP. La CIFP 4, vinculada a la deficiencia y hasta a la ausencia de la proteína de unión 2 (tight junction protein 2, TJP2 por sus siglas en inglés). La CIFP 5 se produce por deficiencia del gen MYO5B (miosina 5 B). Finalmente, la CIFP 6 se origina por la pérdida de función del receptor farnesoide X (farnesoid X receptor, FXR por sus siglas en inglés) a consecuencia de mutaciones en el gen NR1H4. La tres últimas colestasis genéticas se acompañan de GGT normal o disminuida.17, 22
3.4 Errores congénitos de síntesis de ácidos biliares
Los ácidos biliares, uno de los mayores componentes de la bilis, se clasifican en primarios y secundarios. Los primarios son el ácido cólico y quenodeoxicólico, y los secundarios, los ácidos desoxicólico y litocólico. La síntesis de ácidos biliares se realiza a partir del colesterol y se trata de un proceso complejo que necesita diecisiete enzimas que se expresan en el hígado. Son considerados causas raras de CN, que hay que tener en cuenta cuando se han descartado otras causas más frecuentes. Se asocian a defectos moleculares específicos que conducen a la ausencia de ácidos biliares primarios y la concentración de precursores metabólicos hepatotóxicos. El laboratorio se caracteriza por la presencia de colestasis con GGT normal o baja, y ácidos biliares séricos disminuidos. Inicialmente se aconseja realizar cribado de orina con espectrometría de masa y, posteriormente, las técnicas moleculares que permitirán identificar las mutaciones genéticas específicas.2, 3, 22-24
3.5 Fibrosis quística
La enfermedad hepática vinculada con fibrosis quística afecta a menos del 2% de los niños. Debido a esta baja incidencia, el diagnóstico en los pacientes con CN debe estar reservado para aquellos que hayan evidenciado íleo meconial, detención de la curva de peso a pesar del aporte calórico adecuado y aquellos en los cuales otras causas de CN fueron excluidas. El diagnóstico se apoya en la pesquisa neonatal con la determinación de tripsinógeno inmunorreactivo. El estándar de oro es el estudio genético y el test del sudor.2, 3, 17
3.6 Galactosemia
La galactosemia se produce por la incapacidad de metabolización de la galactosa, secundaria a una deficiencia enzimática. La más frecuente es la deficiencia de la galactosa-1-fosfato uridiltransferasa. Es una enfermedad autosómica recesiva, con una incidencia de 1:60 000 nacidos vivos. El defecto metabólico tiene como consecuencia la acumulación de metabolitos tóxicos en el hígado, el cerebro, los riñones y el cristalino ocular. Clásicamente, la enfermedad se presenta a las pocas semanas de vida, luego de que el niño recibe alimentación con leche materna o fórmulas lácteas que contienen lactosa. Los síntomas de presentación incluyen detención del crecimiento, ictericia, vómitos y diarrea. El diagnóstico puede ser sugerido por la presencia de sustancias reductoras en orina; sin embargo, el diagnóstico definitivo necesita la demostración de la ausencia de actividad enzimática, por intermedio de un ensayo realizado en los glóbulos rojos. El tratamiento consiste en la exclusión de alimentos que contienen lactosa y galactosa durante toda la vida.2, 6, 17
3.7 Tirosinemia de tipo 1
La tirosinemia de tipo 1 es una enfermedad metabólica que afecta el metabolismo de los aminoácidos, por deficiencia de la fumarilacetoacetato hidrolasa, enzima responsable del paso final en la degradación de la tirosina. Es una enfermedad autosómica recesiva, con una incidencia de 1:100 000. Puede presentarse en forma aguda durante el período neonatal y se incluye en el diagnóstico diferencial de la insuficiencia hepática aguda neonatal. Puede observarse detención del crecimiento, vómitos, ascitis, coagulopatía, hipoglucemia e hiperbilirrubinemia. El diagnóstico se apoya en la identificación de succinilacetona urinaria elevada.2, 6, 17
4) Colestasis neonatal transitoria
Las causas de CN son muy numerosas y diversas. Los pacientes en los cuales la búsqueda etiológica resulta negativa han sido diagnosticados de causa idiopática, denominada últimamente como “colestasis neonatal transitoria” (5-10% de las CN). Se caracteriza por colestasis de comienzo temprano, ausencia de causas conocidas de CN, normalización clínica y bioquímica durante el seguimiento y el antecedente de algún episodio durante el período neonatal (asfixia, sepsis, nutrición parenteral total, etc).2, 3 A continuación, en la Figura 2, se describe el algoritmo diagnóstico de CN sugerido.
Figura 2. Algoritmo diagnóstico de la CN
Seguimiento de las colestasis neonatales más frecuentes en su evolución a la cronicidad
El objetivo general del seguimiento es lograr que el paciente con patología crónica alcance un óptimo crecimiento y desarrollo, apoyar su nutrición y detectar precozmente las eventuales complicaciones.
En el seguimiento serán pilares fundamentales:
• Educación: los padres deben conocer la enfermedad, el objetivo de los controles y su frecuencia, y deben saber identificar síntomas y signos de alerta con el fin de pesquisar complicaciones en forma precoz. En muchos casos, se los debe preparar para un TH a corto, mediano o largo plazo.
• Detección de complicaciones: dependiendo de la enfermedad de base.
Atresia biliar
Esta colangiopatía es progresiva. El tratamiento quirúrgico inicial, la operación de Kasai, es, en la mayoría de los casos, un puente hacia el TH, de tal manera que al transcurrir los años irán apareciendo las complicaciones y el deterioro progresivo de la función hepática. Estudios multicéntricos han mostrado que alrededor del 98% de los niños operados de AB que viven con su hígado nativo durante más de 5 años tienen manifestaciones clínicas y/o bioquímicas de daño hepático crónico.
Las complicaciones más frecuentes son:
• Colangitis: se presenta en el 60-90% de los casos. A veces es recurrente, conduciendo a un mayor daño hepático. Se vincula al restablecimiento total o parcial del flujo biliar. Se caracteriza por cuadro febril, sin otra causa evidente, y puede asociarse a signos de colestasis como ictericia, acompañado de acolia. El laboratorio presenta un aumento de reactantes de fase aguda, leucocitosis y alteración de las pruebas hepáticas. No se requiere un cultivo positivo confirmatorio. Debe tratarse con antibióticos de amplio espectro. En general se presenta durante los dos primeros años de vida, pero también puede ocurrir posteriormente. El uso profiláctico de antibióticos no es una práctica consensuada y se requieren más estudios que avalen la utilidad de esta conducta. En caso de colangitis recurrente, debe buscarse la presencia de quistes hepáticos como factor determinante de las recurrencias.17, 25
• Hipertensión portal: es otra complicación frecuente en los niños con AB. Se presenta hasta en el 49% de los casos y puede manifestarse con esplenomegalia (56%), hiperesplenismo (43%) (recuento de plaquetas < 150.000), ascitis (17%), síndrome hepatorrenal, hemorragia por várices esofágicas, síndrome hepatopulmonar e hipertensión portopulmonar. Se recomienda la detección de varices esofágicas, para lo cual algunos centros realizan endoscopía digestiva alta después del año, especialmente cuando el recuento de plaquetas es menor de 120.000.2, 17, 26
• Síndrome hepatopulmonar: al igual que otras complicaciones, debe buscarse en forma activa, ya que es poco sintomática. Se presenta en el 10-20% de los niños con hipertensión portal. Se caracteriza por hipoxemia y dilatación microvascular pulmonar. Los niños con AB tienen alto riesgo de desarrollarlo.2, 3, 17, 27
• Hipertensión portopulmonar: es menos frecuente, y también requiere una búsqueda activa para un diagnóstico precoz, ya que en estados avanzados contraindica el TH.8
• Nutrición: en niños con ascitis y gran visceromegalia, la evaluación nutricional será con base en el perímetro braquial y pliegues, ya que el peso no será fidedigno en estos casos. Además, es frecuente ver en estos niños fracturas de huesos largos (8-35%). Esto se relaciona con un déficit y falla en la hidroxilación hepática de la vitamina D.6, 7, 17
• Hepatocarcinoma: aunque es poco frecuente, su asociación con AB puede presentarse en cualquier etapa del seguimiento.8, 28
Síndrome de Alagille
Es una enfermedad multisistémica que requiere un seguimiento multidisciplinario. Uno de los problemas más importantes es el prurito, presente en alrededor del 80% de los casos. Comienza en los primeros meses de vida y a veces es tan mortificante que puede ser una indicación de TH. Otro aspecto típico de esta enfermedad son los xantomas, los que pueden disminuir con la edad o posderivación del flujo biliar. El retraso del crecimiento se ha reportado en el 50-87% de los casos. Las malformaciones vasculares no cardíacas están descritas hasta en un 10%, pudiendo producir accidentes vasculares y hemorragias a distinto nivel y siendo una importante causa de mortalidad. La prevalencia de hepatocarcinoma es baja en relación con las otras enfermedades crónicas; de todas formas, su pesquisa precoz forma parte del seguimiento.2, 6, 29
Déficit de Alfa-1-Antitripsina
Los pacientes PiZZ son los que presentan mayor compromiso hepático (10-15%). La evolución más frecuente es bimodal, presentándose como CN prolongada durante las primeras semanas de vida, mejorando posteriormente. Un 85% tiene pruebas hepáticas normales a los 12 años, manteniendo una buena calidad de vida. Dentro del seguimiento, debe buscarse en forma dirigida el hepatocarcinoma, aunque su frecuencia es baja en la edad pediátrica.18, 19
Colestasis intrahepática familiar progresiva
La identificación del gen involucrado (ATP8B1, ABCB11, ABCB4, TJP2, NR1H4, MYO5B) ha permitido realizar un tratamiento y seguimiento individual de estos pacientes. Los pacientes con CIFP1 (ATP8B1) y CIFP2 (ABCB11) pueden presentar colestasis, prurito y retraso pondoestatural con distintos grados de severidad desde las primeras semanas de vida. En su seguimiento es importante optimizar el manejo nutricional y del prurito. En el caso de la CIFP1 puede agregarse diarrea, insuficiencia pancreática y raquitismo, y en un 80% la talla es inferior al percentilo 3. La indicación de TH no responde solamente al grado de insuficiencia hepática, sino también a la calidad de vida. En el caso de la CIFP2 es frecuente la litiasis biliar y un 15% puede desarrollar hepatocarcinoma o colangiocarcinoma tan precoces como desde los 13 meses de edad. En el resto de las CIFP la incidencia es mucho menor, pero de todas formas es mayor que en la población general. Los pacientes con CIFP3 suelen presentarse más tardíamente, con signos de enfermedad hepática crónica.20-24, 30
Vacunación
Otro aspecto importante en el seguimiento de pacientes con colestasis crónica es monitorizar el programa de inmunizaciones. La morbimortalidad de enfermedades inmunoprevenibles tiene especial importancia en este grupo de pacientes, que en general presenta algún grado de disminución de su inmunidad. Es indispensable que tengan su esquema de vacunación al día, incluyendo vacunas del sarampión, varicela, hepatitis A y B, influenza estacional, neumococo 23V, meningococo tipo B y ACWY (previene la enfermedad meningocóccica causada por los serogrupos A, C, W e Y). En los pacientes candidatos a trasplante se debe considerar adelantar el esquema de vacunación, teniendo en cuenta que el tiempo mínimo requerido para la administración de vacunas con virus vivos y el trasplante es de al menos 4 semanas y 2 semanas para las inactivadas.31
Transición
Finalmente, el seguimiento de los pacientes termina con la “transición a la vida adulta”. El niño debe conocer su enfermedad, sus medicamentos y el equipo médico que lo controla. Es muy importante fortalecer la autonomía y responsabilidad del niño mayor, previniendo la discontinuidad del tratamiento y los controles, que es una causa importante del deterioro de la enfermedad. Por otro lado, es fundamental trabajar en conjunto con los médicos de adultos para que este proceso sea exitoso.2, 3
Tratamiento de la colestasis neonatal
Existen causas de CN que mejoran el pronóstico si son tratadas oportunamente. Cuando no se cuenta con un tratamiento específico, implementar un tratamiento médico inmediato y la optimización de la nutrición son de suma importancia para mejorar la supervivencia, el crecimiento y el desarrollo, así como evitar complicaciones.
Nutrición: la terapia nutricional es esencial para todos los lactantes colestáticos, independientemente de la etiología. En etapas iniciales, los pacientes con grados leves de colestasis o aquellos con portoenteroanastomosis exitosa pueden tener un crecimiento pondoestatural adecuado, mientras reciben lactancia materna o fórmula infantil estándar. Aquellos con colestasis severa requerirán fórmulas infantiles que contengan triglicéridos de cadena media o suplementos con triglicéridos de cadena media, para mejorar la absorción intestinal de lípidos.1, 2
Debido a la presencia de esteatorrea y la mayor demanda metabólica, los pacientes con colestasis requieren un objetivo mínimo para la ingesta de energía de entre 125% y 140% del requerimiento calórico recomendado para la edad. La ingesta proteica puede ser de 2-4 g/kg/día, a menos que exista encefalopatía hepática.1, 2 Cuando los niveles séricos de bilirrubina son > 2 mg/dL, deben suplementarse las siguientes vitaminas liposolubles:
• Vitamina A: 3000-10000 UI/día.
• Vitamina D (colecalciferol): 800-5000 UI/día o 1,25 (OH) 2 colecalciferol: 0.05-0.2 μg/kg/día.
• Vitamina E: 15- 25 UI/kg/día.
• Vitamina K: 2.5-5 mg/día de dos veces por semana a todos los días.
Los niveles séricos de vitaminas y el porcentaje de protrombina deben evaluarse, se ajustarán las dosis de acuerdo con las necesidades específicas de cada paciente.1-3, 6
En pacientes con colestasis crónica, cuando no se logra una ingesta calórica adecuada por vía oral, la alimentación nocturna mediante sonda nasogástrica por goteo o continua, o la nutrición parenteral, pueden ser necesarias para mantener un estado nutricional óptimo. Esto es importante en los candidatos a TH, ya que se ha demostrado que el estado nutricional preoperatorio es un factor pronóstico importante.1-3, 6
Prurito: la causa del prurito en la colestasis no está clara, pero la disminución de los niveles séricos de ácidos biliares ha mostrado una mejoría de los síntomas. Se consideran las siguientes alternativas terapéuticas:
• Ácido ursodesoxicólico: 10-20 mg/kg/día o 600 mg/m2/día, estimula el flujo biliar y, en consecuencia, reduce la gravedad del prurito.
• Rifampicina: 20 mg/kg/día.
• Fenobarbital: 3-10 mg/kg/día.
• Colestiramina: 240 mg/Kg/día, resina quelante de ácidos biliares.
• Hidroxicina (antihistamínico): 2 mg/kg/día.
• Naltrexona: 1-2 mg/kg/día.
• Sertralina: 1-4 mg/kg/día, datos preliminares mostraron que este inhibidor de la recaptación de serotonina es útil en el prurito colestásico refractario en pacientes pediátricos.1-3, 32
Maralixibat y odevixibat son dos drogas aprobadas recientemente por la Food and Drug Administration U.S. (Administración de Alimentos y Medicamentos de EE. UU.) y por la UE (Unión Europea), para tratar el prurito en pacientes con Síndrome de Alagille ≥ 1 año de edad y en CIFP ≥ 3 meses respectivamente. Actúan inhibiendo el transporte de ácidos biliares a nivel ileal, lo que conduce a un aumento de los AB excretados en las heces. Se están realizando investigaciones para sus posibles usos en otras patologías.33, 34
La derivación biliar parcial externa temprana puede representar una opción en niños con colestasis severa asociada a Síndrome de Alagille y CIFP, pudiendo retrasar el TH. Esta técnica fue reportada por Peter Whitington en 1988 y consiste en interponer un asa yeyunal entre la vesícula biliar y la pared abdominal. De esta forma, la interrupción de la circulación enterohepática de los ácidos biliares reduce la cantidad total de estos, lo que resulta útil como tratamiento para el prurito. A pesar de estas medidas, el prurito aún puede ser incontrolable y, en esos casos, el TH puede ser la única opción si la calidad de vida se ve afectada, aun cuando la función hepática sea aceptable.2, 20, 21, 32
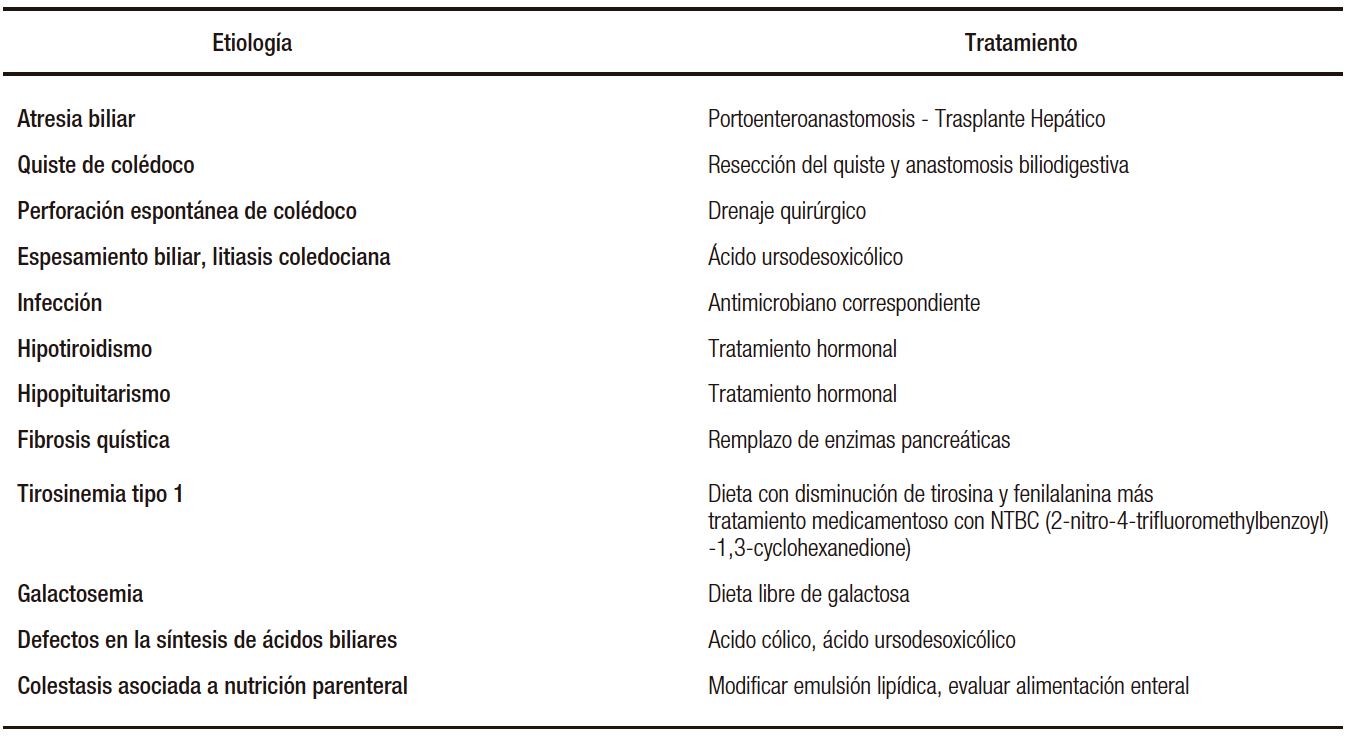
La identificación temprana y el inicio de la terapia para causas tratables de colestasis son cruciales para limitar el daño hepático progresivo y prevenir las lesiones en otros órganos (Tabla 2).
Tabla 2. Etiologías tratables de colestasis neonatal
Trasplante hepático
La indicación de TH y el momento oportuno de su realización dependerá de la etiología y evolución de la CN. Es fundamental valorar los riesgos y beneficios en forma individual para cada paciente, considerando la edad, las enfermedades asociadas, el pronóstico y el entorno biopsicosocial. Los padres y sus familias deben estar preparados con anticipación para este gran paso.1, 2
Conclusiones
La CN es una condición siempre patológica. El diagnóstico precoz permite comenzar en forma temprana y oportuna el tratamiento de aquellas etiologías que responden a un tratamiento específico. Ello determina un importante beneficio para el paciente, ya sea en oportunidad y/o calidad de vida. La AB es reconocida como la causa tratable más frecuente de CN y como la principal causa de trasplante hepático pediátrico en el mundo. Jerarquizar y difundir el conocimiento sobre el diagnóstico de CN, sus etiologías y los estudios y tratamientos posibles podría ofrecer beneficios para los pacientes y la salud pública de los diferentes países, reconociendo la heterogeneidad de situaciones y posibilidades en las diferentes regiones de América Latina.
Propiedad intelectual. Los autores declaran que los datos, las tablas y las figuras presentes en el manuscrito son originales y se realizaron en sus instituciones pertenecientes.
Financiamiento. Los autores declaran que no hubo fuentes de financiación externas.
Conflicto de interés. Los autores declaran no tener conflictos de interés en relación con este artículo.
Aviso de derechos de autor

© 2022 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Godoy M, López C, Álvarez Chavez F y col. Colestasis neonatal: revisión narrativa del grupo de trabajo de la sociedad latinoamericana de gastroenterología, hepatología y nutrición pediátrica. Acta Gastroenterol Latinoam. 2022;52(3):344-354. https://doi.org/10.52787/agl.v52i3.134
Referencias
- Lane E, Murray KF. Neonatal Cholestasis. Pediatr Clin North Am. 2017;64(3):621-39.
- Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, McLin VA, Molleston JP, Neimark E, Ng VL, Karpen SJ. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64(1):154-68.
- Feldman AG, Sokol RJ. Recent developments in diagnostics and treatment of neonatal cholestasis. Semin Pediatr Surg.,2020; 29(4):150945.
- Ananth R. Neonatal Cholestasis: A Primer of Selected Etiologies. Pediatr Ann. 2018;47(11):e433-39.
- Cassoti V, D’Antiga. Liver disease in paedriatric medicine: An overview. In: D’Antiga L. Pediatric Hepatology and liver transplantation. Cham, Switzerland: Springer Nature Switzerland. 2019:3-19.
- Götze T, Blessing H, Grillhösl C, Gerner P, Hoeming A. Neonatal cholestasis – differential diagnoses, current diagnostic procedures, and treatment. Front. Pediatr. 2015;3(43):1-10.
- D’Amato M, Ruiz P, Aguirre K, Gómez S. Cholestasis in Pediatrics. Rev Col Gastroenterol. 2016;31(4):409-17.
- Verkade HJ, Bezerra JA, Davenport M, Schreiber RA, Mieli-Vergani G, Hulscher JB, Sokol RJ, Kelly DA, Ure B, Whitington PF, Samyn M, Petersen C. Biliary atresia and other cholestatic childhood diseases: Advances and future challenges. J Hepatol. 2016;65(3):631-42.
- Schreiber RA: Newborn Screening for Biliary Atresia. JAMA. 2020;323(12):1137-8.
- Jiang J, Wang J, Shen Z, Lu X, Chen G, Huang Y, Dong R, Zheng S. Serum MMP-7 in the Diagnosis of Biliary Atresia. Pediatrics. 2019;144(5):1-9.
- Yang L, Zhou Y, Xu P, Mourya R, Lei H, Cao G, Xiong X, Xu H, Duan X, Wang N, Fei L, Chang X, Zhang X, Jiang M, Bezerra JA, Tang S. Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 for Biliary Atresia. Hepatology. 2018;68:2069-77.
- Gu YH, Yokoyama K, Mizuta K, Tsuchioka T, Kudo T, Sasaki H, Nio M, Tang J, Ohkubo T, Matsui A. Stool color card screening for early detection of biliary atresia and long-term native liver survival: a 19-year cohort study in Japan. J Pediatr, 2015;166:897-902.
- Wolfson JP, Shereiber RA, Butler AE, MacFarlane J, Kaczorowski J, Masucci L, Bryan S, Collet JP. Province wide Biliary Atresia Home screen Program In British Columbia: Evaluation of first 2 years. J Ped Gastroenterol Nutr. 2018;66:845-49.
- Mogul D, Zhou M, Intihar P, Schwarz K, Frock K. Cost Effective Analysis for Screening for Biliary Atresia With the Stool Color Card. J Ped Gastroenterol Nutr. 2015;60:91-8.
- Borgeat M, Korff S, Wildhaber BE. Newborn biliary atresia screening with the stool colour card. BMJ Paediatr Open. 2018;2(1):e000269.
- Martin M, Holmes S, Sim J, Hassan M, Mathew R, Bensen R, Barakat M. Foregone Inclusion: Neonatal CMV Hepatitis and Cholestasis. Dig Dis Sci. 2019;64(11): 3092-5.
- Comité Nacional de Hepatología Pediátrica de la Sociedad Argentina de Pediatría. Consenso de hiperbilirrubinemia del primer trimestre de la vida. Arch Argent Pediatr. 2020;118(1):S12-S49.
- Strnad P, McElvaney NG, Lomas DA. Alpha 1- Antitrypisin Deficiency. N Engl J Med. 2020;382(15):1443-55.
- Cakir M, Sag E, Islek A, Baran M, Tumgor G, Aydogdu S. Liver Involvement in Children whith Alpha-1 Antitrypisin Deficiency: A Multicenter Study. Pediatr Gastroenterol Hepatol Nutr. 2020;23(2):146-53.
- Feldman AG, Sokol RJ. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol. 2019;16(6):346-60.
- Chen HL, Wu SH, Hsu SH, Liou BY, Chen HL, Chang MH. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. 2018;25(1):75.
- Mandato C, Zollo G, Vajro P. Cholestatic jaundice in infancy: struggling with many old and new phenotypes. Ital J Pediatr. 2019;45:83.
- Nicastro E, Di Giorgio A, Marchetti D, Barboni Ch, Cereda A, Iascone M, D’Antiga L. Diagnostic Yield of an Algorithm for Neonatal and Infantile Cholestasis Integrating Next-Generation Sequencing. J Pediatr. 2019;211:54-62.
- Togawa T, Sugiura T, Ito K, Endo T, Aoyama K, Ohashi K, Negishi Y, Kudo T, Ito R, Kikuchi A, Arai-Ichinoi N, Kure S, Saitoh S. Molecular Genetic Dissection and Neonatal/Infantile Intrahepatic Cholestasis Using Targeted Next-Generation Sequencing. J Pediatr. 2016;171:171-7.
- Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic Review: The Epidemiology, Nature History and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-56.
- Henkel SA, Squires JH, Ayers M, Ganoza A, Mckiernan P, Squires JE. Expanding etiology of progressive familial intrahepatic cholestasis. World J Hepatol. 2019;11(5):450-63.
- Seung Hwan B, Ji-Man K, Kyong I, Seok Joo H, Hong K, Jong Gyun A. The Epidemiology and Etiology of Cholangitis After Kasai Portoenterostomy in Patients with Biliary Atresia. J Pediatr Gastroentrol Nutr. 2020;70:171-7.
- Sintusek P, Siriporn N, Punpanich D, Chongsrisawat V, Poovorawan Y. Spleen and Liver Stiffness to Detect Esophageal Varices in Children with Biliary Atresia. J Pediatr Gastroenterol Nutr. 2019;69(4):411-5.
- Snehavardhan P, Khanna R, Lal BB, Sood V, Sood AK, Alam S. Comparison of Two Diagnostic Criteria for Hepatopulmonary Syndrome–High Prevalence in Biliary Atresia. J Pediatr Gastroentrol Nutr. 2020;70:623-7.
- Khanna R, Verma SK. Pediatric hepatocellular carcinoma. World J Gastroenterol. 2018;24(35):3980-99.
- Miller K., Leake K., Sharma T. Advances in vaccinating immunocompomised children. Curr Opin Pediatr. 2020;32(1):145-50.
- Feldman AG, Sokol RJ. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol. 2019;16:346-60.
- Sanchez P, Farkhondeh A, Pavlinov I, Baumgaertel K, Rodems S, Zheng W. Therapeutics Development for Alagille Syndrome. Front. Pharmacol. 23 de Agosto de 2021 | https://doi.org/10.3389/fphar.2021.704586
- Deeks ED. Odevixibat: First Approval. Drugs. 2021;81:1781-6.
Correspondencia: Marcela Godoy
Correo electrónico: mmgodoyp@gmail.com
Acta Gastroenterol Latinoam 2022;52(3):344-354
