Alba Santiago Badenas ID· Elena F Verdu ID
Farncombe Family Digestive Health Research Institute, Department of Medicine, McMaster University, Hamilton, Canadá.
Acta Gastroenterol Latinoam 2021;51(4):385-393
Recibido: 16/09/2021 / Aceptado: 20/10/2021 / Publicado en www.actagastro.org el 13/12/2021 / https://doi.org/10.52787/ZDTT9566

Resumen
La enfermedad inflamatoria intestinal es una afección inmunomediada que incluye la enfermedad de Crohn y la colitis ulcerosa y causa inflamación crónica del tracto gastrointestinal. Aunque se desconoce la causa exacta de la enfermedad inflamatoria intestinal, existe consenso acerca de que una combinación de factores genéticos, ambientales e inmunitarios participan en su patogenia. A lo largo de todos estos años se han enfatizado las alteraciones que ocurren una vez desarrollada la enfermedad; sin embargo, aún no están claras las causas que la desencadenan. Existe una base genética evidente en la que se ven afectados genes implicados en la homeostasis intestinal, tales como NOD2, ATG16L1 o XBP1. Sin embargo, estos factores genéticos no son suficientes y se necesitan factores ambientales adicionales e interacciones alteradas entre la microbiota intestinal y el sistema inmunológico. Una menor diversidad microbiana y una disminución en la abundancia relativa de Firmicutes, así como un aumento de Proteobacteria, se describen como los cambios más consistentes en la composición microbiana entre los pacientes con enfermedad inflamatoria intestinal. Además de las variaciones en la composición, también se han observado cambios funcionales en estudios transversales. No obstante, la adición de cohortes longitudinales nos permite identificar comunidades y funciones microbianas específicas que podrían estar influyendo en la aparición de la enfermedad. Así, el estudio realizado en una cohorte de individuos en riesgo de desarrollar la enfermedad inflamatoria intestinal (cohorte GEM) mostró un aumento de la actividad proteolítica fecal, asociada a cambios en la composición microbiana, antes del inicio de la colitis ulcerosa. Estos hallazgos podrían ayudar a desarrollar nuevas técnicas de diagnóstico no invasivas, así como nuevos enfoques terapéuticos.
Palabras claves. Enfermedad inflamatoria intestinal, microbiota intestinal, microbioma previo a la enfermedad, actividad proteolítica intestinal.
Emerging Pathogenic Mechanisms in Inflammatory Bowel Disease: Role of Bacterial Proteolytic Activity
Summary
Inflammatory bowel disease is an immune mediated condition that includes Crohn’s disease and ulcerative colitis and causes chronic inflammation of the gastrointestinal tract. Although the exact cause for inflammatory bowel disease is unknown, there is consensus that a combination of genetic, environmental, and immune factors participate in its pathogenesis. To date, all the studies have been focused on alterations that occur once IBD has developed, however, the causes triggering the onset of the disease are still unclear. There is an evident genetic basis in which multiple genes involved in intestinal homeostasis are affected, such as NOD2, ATG16L1 and XBP1. However, these genetic factors are not sufficient for disease onset and additional environmental factors such as those related to dysregulation of gut microbiota and the immune system are required. A lower microbial diversity and a decrease in the relative abundance of Firmicutes, as well as an increase in Proteobacteria, have been described in patients with inflammatory bowel disease, but are not found in all studies. In addition to variations in microbial composition, functional changes have also been observed in cross-sectional studies. Longitudinal cohorts in patients at risk for inflammatory bowel disease have recently been conducted allowing us to interrogate whether specific microbial communities and functions could be influencing the onset of the disease. Indeed, a translational study performed in a cohort of at-risk individuals for inflammatory bowel disease (GEM cohort) showed an increased fecal proteolytic activity, associated with microbial composition changes, before the onset of ulcerative colitis. These findings may help develop new non-invasive diagnostic techniques, as well as new therapeutical approaches for inflammatory bowel disease.
Keywords. Inflammatory bowel disease, intestinal microbiota, pre-disease microbiome, intestinal proteolytic activity.
Abreviaturas
EII: Enfermedad inflamatoria intestinal.
EC: Enfermedad de Crohn.
CU: Colitis ulcerosa.
IBD: Inflammatory bowel disease.
CD: Crohn’s disease.
UC: Ulcerative colitis.
ROS: Especies reactivas de oxígeno.
SNPs: Polimorfismos de nucleótido único.
PRR: Receptor de reconocimiento de patrones.
MUC: Mucinas.
NK: Células natural killer.
DC: Células dendríticas.
PAMP: Patrones moleculares asociados a patógenos.
IFN: Interferón.
CMH: Complejo mayor de histocompatibilidad.
AINEs: Analgésicos antinflamatorios no esteroideos.
AGCC: Ácidos grasos de cadena corta.
GWAS: Estudio de asociación de genoma completo.
Introducción
La enfermedad inflamatoria intestinal (EII) incluye la colitis ulcerosa (CU) y la enfermedad de Crohn (EC), ambas caracterizadas por inflamación crónica del tracto digestivo, con períodos de exacerbación y de etiopatogenia compleja.1 Aunque en las últimas décadas la incidencia de la EII en el mundo occidental se ha estabilizado, oscilando entre 10-30 por cada 100.000, la prevalencia sigue siendo elevada, afectando hasta a un 0,5% de la población global.2 Se ha observado una tendencia al alza de la incidencia en aquellos países recientemente industrializados. Estas observaciones sugieren que a medida que los países en desarrollo se occidentalicen, las tasas de prevalencia e incidencia de la EII deberían crecer en paralelo a las del mundo occidental. Además, también se ha evidenciado cómo la descendencia de inmigrantes provenientes de países con baja incidencia acaba teniendo un riesgo similar a desarrollar EII que la población base del país desarrollado, pudiendo estar relacionado con la adopción de un nuevo estilo de vida y hábitos alimentarios.2, 3
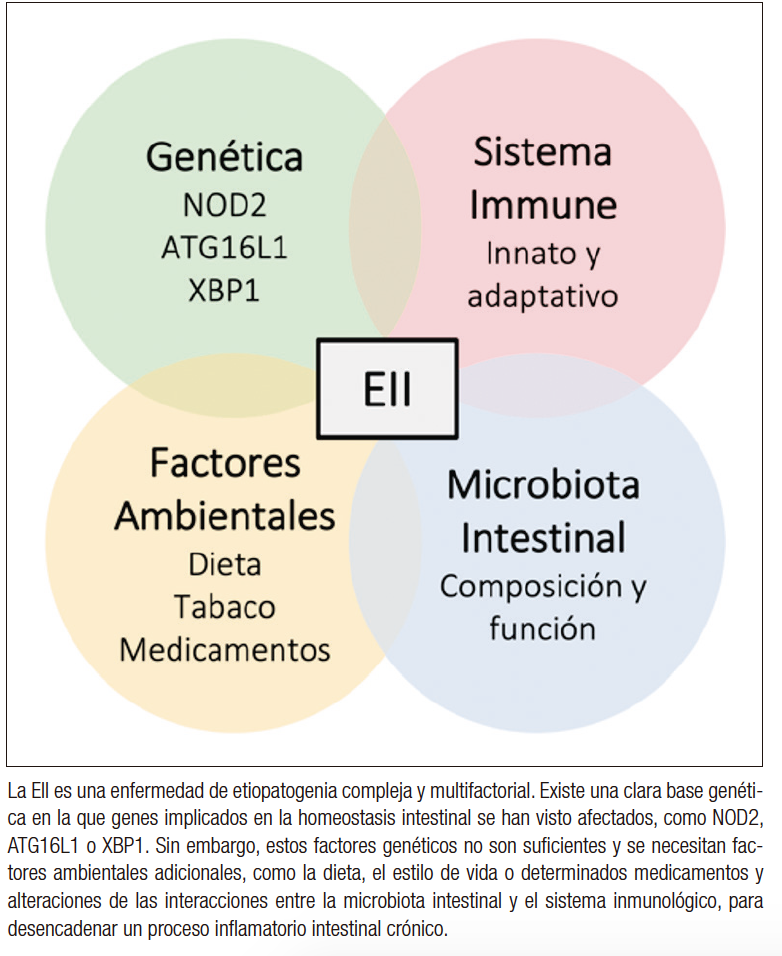
Aunque ambas formas principales de EII muestran características clínicas y patológicas similares, la CU se caracteriza por una inflamación difusa y continua que puede afectar a la mucosa del recto y colon, mientras que la EC se caracteriza por inflamación transmural, de forma discontinua, o en forma de parches, que puede afectar a todo el tracto gastrointestinal, siendo más común en el colon y el íleon terminal.4 Se desconoce la causa exacta de la EII, pero existe consenso para describirla como una enfermedad de etiología multifactorial en la que factores genéticos, inmunológicos y ambientales, incluyendo alteraciones de la microbiota intestinal, estarían involucrados en su patogénesis (Figura 1).1, 5
Figura 1. Factores involucrados en el desarrollo de la EII
A lo largo de todos estos años se han enfatizado las alteraciones que ocurren una vez desarrollada la enfermedad; sin embargo, aún no están claras las causas que la desencadenan. Por ello, esta revisión trata de destacar los hallazgos mostrados en los diferentes estudios transversales y longitudinales publicados hasta el momento, para poder esclarecer los factores que podrían llevar a la prevención de la EII, con particular enfoque en el microbioma intestinal y su actividad proteolítica.
Fisiopatología
La susceptibilidad a la EII posee una clara base genética, fundamentada en estudios poblacionales en los que se ha demostrado que existe una elevada agregación familiar en la enfermedad; donde el riesgo de padecer CU o EC en familiares de probandos está aumentado de 8 a 10 veces.6 Además, estudios de concordancia gemelar también proporcionan evidencia de la predisposición genética, jugando un papel menor en CU en comparación con EC.7 Tras la descripción del primer gen de susceptibilidad, el NOD2 en la EC,8 y gracias a los estudios de asociación de todo el genoma (GWAS), hasta la fecha, se han podido identificar más de 230 loci de riesgo genético de la EII.5, 9 El análisis de los genes y los loci genéticos implicados en la EII han ayudado a comprender la importancia de ciertas vías en la homeostasis intestinal, incluidas la función de barrera epitelial, la defensa frente a microorganismos, la regulación inmunitaria, la migración celular, la generación de especies reactivas de oxígeno (ROS), la autofagia y vías metabólicas asociadas con la homeostasis celular, entre otras.10, 11
Se sabe que la barrera intestinal es una importante interfaz física, química e inmunológica entre la microbiota intestinal y el huésped; así, cualquier desregulación de esta barrera podría contribuir al desarrollo de la enfermedad. Entre algunos de los genes descritos asociados al riesgo de EII se encuentran DLG5, MUC19, PTGER4 e ITLN1.5, 11-14 Entre los genes implicados en el reconocimiento de antígenos microbianos, encontramos el NOD2, descrito como el loci de riesgo más fuerte en la EII,5, 15 aunque también podemos encontrar otros involucrados como CARD9.10 Otros de los genes importantes descritos gracias a los estudios genéticos han sido ATG16L116 e IRGM,17 ambos relacionados con la autofagia, y XBP1, componente clave del estrés del retículo endoplasmático;18 dos procesos que han demostrado tener también un papel importante en las respuestas inmunitarias en la EII. Cabe destacar también la identificación de diversos polimorfismos de nucleótido único (SNPs) en IL-23R, con alta asociación para la EII, debido a su papel en la activación de linfocitos y la producción de citoquinas. El gen IL23R codifica una subunidad del receptor de la citoquina IL-23 involucrada en la diferenciación de la línea celular Th17, encargada de la producción y liberación de citoquinas proinflamatorias como IL17.19
Sin embargo, dado que los factores genéticos representan solo una parte de la variación general de la enfermedad, se requieren factores ambientales adicionales y alteraciones de las interacciones entre la microbiota intestinal y el sistema inmunológico para el desarrollo de la EII. Precisamente, de los genes mejor caracterizados con relación a EII, el NOD2, que es un receptor de reconocimiento de patrones (PRR) intracelular, desempeña un papel esencial en el mantenimiento del equilibrio entre la microbiota y las respuestas inmunitarias del huésped. Se expresa tanto en células del sistema inmune innato como en células epiteliales y reconoce componentes de la pared celular bacteriana. Su activación promueve la eliminación de patógenos mediante la activación de un programa transcripcional proinflamatorio y de otras vías inmunitarias innatas.20, 21 Este descubrimiento llevó a un cambio de enfoque, desde estudios centrados en el sistema inmunitario adaptativo hacia la importancia del sistema inmunitario innato y, más específicamente, su rol en la detección de componentes microbianos en el lumen intestinal.22
La barrera epitelial intestinal representa la primera línea de defensa contra una infección. Defectos en esta barrera se han descrito en la EII, destacando un aumento de la permeabilidad debido a una alteración en la expresión de las uniones estrechas, facilitando así una mayor exposición antigénica y, con ello, una activación inmunitaria.23, 24 Además, se ha demostrado la importancia de la capa mucosa, compuesta principalmente de glicoproteínas ramificadas, incluidas las mucinas (MUC), en la prevención de la penetración bacteriana y la inflamación intestinal. Este rol se ha puesto de manifiesto mayoritariamente en estudios llevados a cabo en modelos de ratones knock out para MUC2 (MUC2−/−) con colitis.25-27
Una segunda capa crucial de protección inmunológica intestinal se basa en una respuesta rápida y eficaz contra los microorganismos que logran penetrar más allá de las células epiteliales. Esto ocurre a través del sistema inmune innato y mediado por una gran variedad de tipos de células, incluidos los neutrófilos, macrófagos, monocitos, células dendríticas y células natural killer (NK). Esta respuesta puede estar mediada por mecanismos dependientes de células, como fagocitosis, o por factores secretados.28 Esta respuesta se inicia cuando los PRR, expresados no solo en macrófagos y células dendríticas (DC) sino también en diversas células no inmunes, permiten el reconocimiento de estructuras conservadas entre las especies microbianas, denominadas patrones moleculares asociados a patógenos (PAMP). Estos PRR activan una cascada que resulta en la transcripción de genes implicados en la respuesta inflamatoria, que codifican citoquinas proinflamatorias, interferones tipo I (IFN), proteínas antimicrobianas y quimiocinas, y que además permiten el reclutamiento de neutrófilos al lugar de la inflamación.29 Sin embargo, alteraciones en el reconocimiento de patógenos intracelulares, como las asociadas a polimorfismos de NOD2, podrían conllevar a respuestas inmunológicas innatas inadecuadas, al alterar el reconocimiento de PAMPs y disminuir el aclaramiento de patógenos, conduciendo así a una inflamación aberrante.21, 30 Además, también se ha descrito que variantes de NOD2 podrían influir en una producción defectuosa de péptidos antimicrobianos, como las α-defensinas, por parte de las células de Paneth.31, 32 Pero aparte de su papel principal en la inmunidad innata, también es capaz de activar el sistema inmunitario adaptativo. Por lo tanto, estos polimorfismos podrían producir un defecto en la inmunidad innata impidiendo generar una respuesta inflamatoria adecuada para controlar la microbiota y coordinar la respuesta inmunitaria adaptativa, favoreciendo así un proceso inflamatorio crónico.20, 23
Mientras que el sistema inmune innato induce inicialmente la respuesta inflamatoria, el sistema inmune adaptativo es el que juega un papel central en la progresión de los eventos inflamatorios crónicos observados en la EII.33 Los principales efectores del sistema inmune adaptativo son los linfocitos B y T, que confieren respuestas inmunes específicas y que pueden tardar varios días en responder. Los linfocitos B, efectores de la respuesta humoral, son los encargados de secretar inmunoglobulinas, como la IgA o IgG.33 No obstante, los linfocitos T son la población de células clave de esta inmunidad adaptativa.34 Estos son activados una vez que reciben señales derivadas de las células presentadoras de antígeno, como las DC, a través del complejo mayor de histocompatibilidad (CMH) y diferenciados en una subpoblación de células T diferente en función de la presencia de distintas citoquinas.35 Inicialmente, se pensaba que la EC se caracterizaba por una respuesta inmune mediada por las células Th1, que secretan IFNγ e IL-2, mientras que la CU se consideraba una enfermedad mediada por células Th2, que secretan IL-4, IL-5 e IL-13, entre otras.34, 36 En las últimas décadas, se ha descubierto que otro tipo celular, las células Th17, también están implicadas en la patogénesis de la EII. Estas células, que producen IL-17A, IL-17F, TNFα e IL-22, entre otras, parece ser que jugarían un papel importante durante la inflamación intestinal crónica, ya que se ha visto que están involucradas en el daño tisular.36-38
El importante papel que juegan los factores ambientales en la patogenia de la EII está respaldado por el patrón epidemiológico de esta, en el que se ha observado un aumento de la prevalencia en países desarrollados, con una tendencia al alza en la incidencia de la enfermedad en países recientemente industrializados.2, 3 Por tanto, existe una serie de factores medioambientales que van desde la concepción, pasando por la infancia y la posterior exposición asociados a la vida moderna, como la dieta, el uso de antibióticos, tabaquismo, los cambios en la exposición a microorganismos ambientales, el estrés social, etc., que influyen en el riesgo de desarrollar la enfermedad.22, 39, 40 El tabaquismo se considera uno de los factores más significativos que alteran el curso clínico de la EII. Mientras que el tabaco empeora muchos aspectos de la EC, en la CU parece tener efectos protectores.41 La dieta también es considerada un factor ambiental importante que afecta el desarrollo de la EII. Los estudios han demostrado que la ingesta de frutas y verduras está asociada con una disminución del riesgo de EII, mientras que la ingesta de comidas rápidas, ricas en grasa, sobre todo animal, y azúcares podría aumentarlo. Los niveles bajos de vitamina D también parecen ser un factor de riesgo para la EII. Además, últimamente, se está señalando a los aditivos alimentarios artificiales como posibles promotores de inflamación intestinal, al interferir con la función de barrera en el intestino y la interacción huésped-microbiota.42-44 El uso de medicamentos como los AINEs o los antibióticos también son un factor a tener en cuenta, siendo estos últimos un factor que puede afectar desde la infancia. Varios estudios han demostrado que el uso de antibióticos en niños parece aumentar el riesgo de desarrollar EII en población pediátrica.40
Muchos de estos factores actúan en conjunto modificando no solo la composición de la microbiota intestinal, sino también su función. La microbiota intestinal, que podría sumar hasta cien billones de células, diez veces el número de células humanas, es esencial para la salud del huésped y desempeña un rol importante en muchos aspectos de la fisiología humana, desde procesos simples hasta complejos, como metabolismo, resistencia a patógenos o en la maduración y regulación del sistema inmunitario, entre otros.45-48 Así, las implicaciones funcionales de la disbiosis, descrita como una desviación en la composición y función de la microbiota intestinal,49 pueden afectar la fisiología humana, el estado de salud y la susceptibilidad a enfermedades, entre ellas la EII. Sin embargo, queda mucho para determinar si la disbiosis es un factor contribuyente o una consecuencia de la enfermedad.50
Alteraciones de la microbiota en la EII
Como se ha descrito anteriormente, la EII es una enfermedad de etiología compleja en la que individuos genéticamente predispuestos y bajo la influencia de factores ambientales desencadenarían una respuesta inmune aberrante contra microbios intestinales, perpetuando una inflamación crónica del tracto gastrointestinal.51
En la EII se han descrito cambios microbianos tanto en su composición como funcionales.52, 53 A grandes rasgos, los cambios más consistentes observados en pacientes con EII han sido una diversidad microbiana más baja y una disminución en la abundancia relativa de Firmicutes. Además, también se ha descrito un aumento de proteobacteria, con predominio de Enterobacteriaceae.54, 55 Varios estudios coinciden en que existe una abundancia reducida de múltiples especies de bacterias productoras de butirato, entre ellas Faecalibacterium prausnitzii y Roseburia.56, 57 Otro de los hallazgos principales en esta forma de EII es el aumento de la Escherichia coli invasora-adherente (proteobacteria),58, 59 así como un aumento de bacterias mucolíticas, como Ruminococcus gnavus.60, 61 Se han descrito otros cambios taxonómicos específicos, pero la interpretación de estos es compleja y no siempre concordante, debido a la heterogeneidad de los estudios, con poblaciones, muestras y procesamiento de estas muy dispares. Un claro ejemplo es la controversia en la asociación de la abundancia relativa de bacteroides y la EII; mientras que varios estudios observan una disminución de esta población en enfermedad,56, 62, 63
otros estudios describen un aumento en pacientes con EII activa,52, 56 además del rol de B. vulgatus en la exacerbación de la colitis experimental.64 Esto no es sorprendente teniendo en cuenta que Bacteroides es un taxón muy diverso y, precisamente, la controversia reafirma la importancia de entender mecanismos asociados a especies bacterianas.
Por otro lado, la CU se asocia también con una disminución de la diversidad microbiana; en cambio, parece ser que no habría una disbiosis tan marcada como en la EC, e incluso se habría observado una semejanza a nivel de composición taxonómica más similar a controles sanos que a EC.54, 65 Aun así, también se ha descrito una disminución de la abundancia relativa de bacterias productoras de butirato como F. prausnitzii tanto en pacientes con CU activa66 como con CU en remisión, viéndose una recuperación gradual de estas bacterias en aquellos pacientes con remisión prolongada,67 así como de Roseburia.68 Varios estudios también han demostrado el posible efecto protector de Akkermansia muciniphila, cuya presencia se ve disminuida en pacientes con CU.61, 69
Además de las variaciones en la composición microbiana, también se han observado cambios funcionales. De manera general, se ha observado una disminución de la “diversidad metabólica” entre los pacientes con EII, análoga a la disminución de diversidad en la composición.60 Como cambios funcionales destacables en pacientes con EII, se han descrito concentraciones fecales reducidas de ácidos grasos de cadena corta (AGCC), como el butirato, cambios en las vías del estrés oxidativo, disminución del metabolismo de carbohidratos y disminución de la biosíntesis de aminoácidos.56, 70, 71
Lecciones de los estudios GEM
La mayoría de los estudios han examinado a pacientes con enfermedad activa o tratada utilizando un diseño de casos y controles de corte transversal, es decir, después de que la inflamación y la lesión tisular ya se han desarrollado, lo que dificulta discernir si los cambios descritos son causa o consecuencia de la inflamación. Por otro lado, se sabe poco acerca de los cambios funcionales o de composición microbiana que ocurren antes del inicio de la enfermedad. Por ello, se llevó a cabo un estudio usando la cohorte GEM, una cohorte longitudinal de pacientes en riesgo de EC, diseñada para investigar las interacciones genéticas, ambientales y microbianas que podrían causar la enfermedad. Inesperadamente, algunos de los sujetos incluidos en el estudio desarrollaron CU en lugar de EC. Así, usando este subconjunto único de muestras, se investigó si funciones microbianas específicas podrían estar influyendo en el inicio de la enfermedad.55
De acuerdo con los estudios transversales previamente descritos, en esta cohorte prospectiva también se observaron cambios en la composición microbiana, siendo más evidentes en aquellas muestras de pacientes una vez desarrollada la enfermedad (post-CU) en comparación con las muestras previas al desarrollo de la enfermedad (pre-CU) o las de los controles. Entre ellos, cabe destacar una disminución en la diversidad microbiana, así como de la abundancia de bacterias beneficiosas, como Adlercreutzia y Bifidobacterium, y mayor abundancia de patógenos, como Actinobacillus. En este estudio, además de las asociaciones basadas en anomalías estructurales de la microbiota con estados patológicos, también se quiso enfatizar la importancia de las contribuciones funcionales de la microbiota. De los más de 20.000 genes bacterianos que fueron significativamente diferentes entre los tres grupos, cabe destacar que un importante número de estos se relacionaron con proteasas y peptidasas, permitiendo una diferenciación clara entre pre-CU y controles.55
El equilibrio proteolítico se ha descrito como una de las posibles contribuciones funcionales de la microbiota. El tracto gastrointestinal está expuesto a diferentes proteasas, que tienen funciones claves en procesos como la digestión, la inmunidad y la función de barrera. En la EII se ha descrito un desequilibrio en la función proteolítica caracterizado por un aumento de las proteasas y/o una disminución de las antiproteasas del huésped, como el inhibidor de serina proteasa (serpina), llamado elafina.72, 73 Sin embargo, las bacterias, incluidas las presentes en el intestino humano, también son una fuente conocida de proteasas, muchas de las cuales son proinflamatorias.74 Teniendo en cuenta esto, y junto con los hallazgos en la expresión génica de la muestras pre-CU, se conjeturó que los cambios en la actividad proteolítica microbiana intestinal en pre-CU podrían representar un mecanismo por el cual las proteasas derivadas de la microbiota contribuyen al inicio espontáneo de la inflamación. Así, se observó que tanto la actividad proteolítica total como la actividad elastolítica estaba aumentada en aquellas muestras pre-CU, llegando a niveles similares a los observados en post-CU. Además, la actividad elastolítica se correlacionó inversamente con taxones bacterianos beneficiosos como Adlercreutzia o Akkermansia,69, 75 mientras que se correlacionó positivamente con Bacteroides vulgatus. Sugiriendo, así, un componente microbiano de la actividad proteolítica.
La colonización de ratones libres de gérmenes (germ free), tanto en adultos como en modelo neonatal, con heces de pacientes pre-CU y post-CU, permitió confirmar una transferencia del fenotipo proteolítico, con el consiguiente efecto proinflamatorio en los ratones.55 Esta activación inmune a través de proteasas microbianas ya se había descrito previamente en el intestino delgado in vivo.76
Como se ha descrito anteriormente, la EII es una enfermedad de fisiopatología compleja en la que hay una relación multidireccional entre los factores ambientales, genéticos, microbianos e inmunológicos. Entre ellos, la microbiota intestinal es la que ha adquirido un rol central en los últimos tiempos como principal desencadenante de una respuesta inmune aberrante. Hasta ahora se había descrito qué es lo que estaba sucediendo una vez establecida la enfermedad, pero los estudios longitudinales como el aquí descrito, llevado a cabo por Galipeau et al.,55 nos permitirán ir más allá y ser capaces de describir qué sucede justo antes del inicio de la enfermedad, no solo a nivel composicional sino también funcional, y de esta manera poder desarrollar tanto nuevas técnicas no invasivas de diagnóstico como nuevos métodos de abordaje terapéutico.
Conclusiones y futuro
La base microbiana en la EII se ha planteado desde principios del siglo xx77 y tiene hoy en día un rol central en su fisiopatología, considerándose el principal factor ambiental desencadenante de inflamación crónica. Además del estudio de la composición microbiana, el estudio funcional, a través de la producción de metabolitos que interactúan con la barrera intestinal y el sistema inmune, y la digestión de productos de la dieta, han cobrado un interés central en los últimos años. El desafío consiste en establecer causalidad e identificar taxones bacterianos y vías patogénicas que permitan desarrollar nuevos enfoques terapéuticos. Para ello se requiere cooperación entre la investigación básica, clínica y traslacional.
Propiedad intelectual. Los autores declaran que los datos y las figuras presentes en el manuscrito son originales y fueron realizados en las instituciones a las que pertenecen.
Aviso de derechos de autor

© 2021 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Badenas AS, Verdu EF. Mecanismos patogénicos del microbioma en la enfermedad inflamatoria intestinal: rol de la actividad proteolítica bacteriana. Acta Gastroenterol Latinoam. 2021;51(4):385-3 https://doi.org/10.52787/ZDTT9566
Referencias
- Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nature Reviews Gastroenterology and Hepatology 2015. https://doi.org/10.1038/nrgastro.2015.34.
- Kaplan GG. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015;12:720-7.
- Ng SC, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769-78.
- Baumgart DC, WJS. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet, London. 2007;369:1641-57.
- Turpin W, Goethel A, Bedrani L, Croitoru K. Determinants of IBD Heritability: Genes, Bugs, and More. Inflamm. Bowel Dis. 2018;24:1133-48.
- Loddo I, Romano C. Inflammatory bowel disease: Genetics, epigenetics, and pathogenesis. Front. Immunol. 2015;6.
- Halfvarson J. et al. Longitudinal concordance for clinical characteristics in a Swedish-Danish twin population with inflammatory bowel disease. Inflamm. Bowel Dis. 2007;13:1536-44.
- Hugot J. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599-603.
- De Lange KM, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49:256-61.
- Khor B, Gardet A, RJX. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature 2011;474:307-17.
- Mirkov MU, Verstockt B, Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol. Hepatol. 2017;2:224-34.
- Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955-62.
- Stoll M, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476-80.
- Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn’s disease. Annu. Rev. Genomics Hum. Genet. 2009;10:89-116.
- Inohara N, Nuñez G. NODS: Intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 2003;3:371-82.
- Rioux JD, et al. Genome-wide association study identifies five novel susceptibility loci for Crohn’s disease and implications a role for autophagy in disease pathogenesis. Nat. Genet. 2007;39:596-604.
- Mccarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. 2008;40:1107-12.
- Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743-56.
- Richard H., Duerr Kent D, Taylor Steven R, Brant John D, Rioux Mark S, Silverberg Mark J, Daly A Hillary Steinhart, Clara Abraham, et al. A Genome-Wide Association Study Identifies IL23R as an Inflammatory Bowel Disease Gene. Science (80-. ). 314, 1461–1463 (2006).
- Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: current insights. J. Inflamm. Res. 2018;11:49-60.
- Watanabe T, Kitani A, Strober W. NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn’s disease. Gut 54. 2005;1515-18.
- Zhang YZ, Li YY. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014;20:91-9.
- Salim SY, Söderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011;17:362-81.
- Choy MC, Visvanathan K, De Cruz P. An overview of the innate and adaptive immune system in inflammatory bowel disease. Inflamm. Bowel Dis. 2017;23:2-13.
- Van der Sluis M, et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That MUC2 Is Critical for Colonic Protection. Gastroenterology. 2006;131:117-29.
- Larsson JMH, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011;17:2299-307.
- Johansson MEV, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013;10:352-61.
- Gasteiger G, et al. Cellular Innate Immunity: An Old Game with New Players. J. Innate Immun. 2017;9:111-25.
- Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140:805-20.
- Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity. 2014;41:898-908.
- Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol. 2011;4:484-95.
- Sidiq T, Yoshihama S, Downs I Kobayashi KS. Nod2: A critical regulator of ileal microbiota and Crohn’s disease. Front. Immunol. 2016;7:18-20.
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007:448;427-34.
- Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014;13:3-10.
- Zhu J, PWE. Peripheral CD4 T cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247-62.
- Zenewicz L.A, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol. Med. 2009;15:199-207.
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol. Rev. 2008;223:87-113.
- Steinman L. A brief history of TH17, the first major revision in the T H1/TH2 hypothesis of T cell-mediated tissue damage. Nat. Med. 2007;13:139-45.
- Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012;9:599-608.
- Ananthakrishnan AN, et al. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018;15:39-49.
- Birrenbach T, BU. Inflammatory bowel disease and smoking. Inflamm. Bowel Dis. 2004;10:848-59.
- Dolan KT, Chang EB. Diet, gut microbes, and the pathogenesis of inflammatory bowel diseases. Mol Nutr Food Res. 2017;61:1-20.
- Lewis JD, Abreu MT. Diet as a trigger or therapy for inflammatory bowel diseases. Gastroenterology. 2017;152:398-414.
- Bancil AS, et al. Food additive emulsifiers and their impact on gut microbiome, permeability, and inflammation: mechanistic insights in inflammatory bowel disease. J. Crohns. Colitis. 2021;15:1068-79.
- Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing : Article : Nature. Nature. 2010;464:59-65.
- Gerritsen J, Smidt H, Rijkers GT, De Vos WM. Intestinal microbiota in human health and disease: The impact of probiotics. Genes Nutr. 2011;6:20940.
- Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189-200.
- Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. 2012;489:23141.
- Lynch SV, Pedersen O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016;375: 2369-79.
- Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Heal. Dis. 2015;26:1-9.
- Turpin W, et al. Increased Intestinal Permeability Is Associated With Later Development of Crohn’s Disease. Gastroenterology; 159:2092-100.
- Lloyd-Price J, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:65562.
- Vich Vila A, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018;10:1-11.
- Pascal V, et al. A microbial signature for Crohn’s disease. Gut. 2017;66:81322.
- Galipeau H J, et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology. 2021;160:153245.
- Wright EK, et al. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: A systematic review. Inflamm. Bowel Dis. 2015;21:1219-28.
- Takahashi K. et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion. 2016;93:59-65.
- Baumgart M, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1:403-18.
- Martinez-Medina M, et al. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009;15:87282.
- Franzosa EA, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019;4:293-305.
- Png CW, et al. Mucolytic Bacteria With Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. 2010;105:2420-28.
- Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza YV, Treuren W, Ren B, Schwager E, Knights D, et al. The treatment-naïve microbiome in new-onset Crohn’ s disease. Cell Host Microb. 2014;15:38292.
- Takaishi, H. et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int. J. Med. Microbiol;298:46372.
- Bloom SM, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390-403.
- Sankarasubramanian J., Ahmad R, Avuthu N, Singh AB, Guda C. Gut microbiota and metabolic specificity in ulcerative colitis and crohn’s disease. Front. Med. 2020;7:1-15.
- Sokol H. et al. Low counts of faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009;15:1183-9.
- Varela E, et al. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013;38:151-61.
- Machiels K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacteriumprausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 63;1275-83.
- Earley H, et al. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci. Rep. 2019;9:1-9.
- Huda-Faujan N, Abdulamir AS, Fatimah AB, Muhammad Anas O, et al. The Impact of the level of the intestinal short chain fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem. J. 2010;4: 53-8.
- Marchesi JR, et al. Rapid and Noninvasive Metabonomic Characterization of Inflammatory Bowel Disease research articles. 2007:546-51.
- Vergnolle N. Protease inhibition as new therapeutic strategy for GI diseases. Gut. 2016;65:1215-24.
- Motta J, et al. Modifying the protease, antiprotease pattern by Elafin overexpression protects mice from colitis. Gastroenterology. 2011;140:1272-82.
- Carroll IM, Maharshak N. Enteric bacterial proteases in inflammatory bowel diseasepathophysiology and clinical implications. World J. Gastroenterol. 2013;19:7531-43.
- Illescas O, Rodríguez-Sosa M, Gariboldi M. Mediterranean diet to prevent the development of colon diseases: A meta-analysis of gut microbiota studies. Nutrients. 2021;13:1-21.
- Caminero A, et al. Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat. Commun. 2019;10:1-14.
- Hawkins HP. An address on the natural history of ulcerative colitis and its bearing on treatment. Br. Med. J. 1909;1:765-70.
Correspondencia: Alba Santiago Badenas
Correo electrónico: santiaga@mcmaster.ca
Acta Gastroenterol Latinoam 2021;51(4):385-393
