
María Virginia D´Ascenzo,1 José Daniel Bosia,1 Silvia Mabel Borzi,1 Ezequiel Barán,2 Gabriel Ricardo García,2 María Cecilia Calzona1
1 Servicio de Gastroenterología.
2 Servicio de Neumonología.
Unidad de Fibrosis Quística de Adultos. Hospital Profesor Rodolfo Rossi. Ciudad de La Plata, provincia de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2020;50(1):9-19
Recibido: 26/11/2018 / Aceptado: 16/05/2019 / Publicado en www.actagastro.org el 23/03/2020 / https://doi.org/10.52787/vipw2810

Resumen
Introducción. El incremento en la sobrevida de pacientes adultos con fibrosis quística (FQ) ha generado una mayor relevancia de la enfermedad extrapulmonar. Objetivo. Determinar el compromiso digestivo, nutricional y el impacto en la sobrevida de pacientes adultos con FQ. Métodos. Estudio retrospectivo, descriptivo y comparativo de 100 pacientes adultos con diagnóstico de FQ, asistidos entre 2003 y 2017. Se valoraron parámetros clínicos, genéticos, índice de masa corporal (IMC), pruebas de función respiratoria, estudio bacteriológico de esputo, insuficiencia pancreática (IP), diabetes relacionada con FQ (DRFQ), pancreatitis aguda (PA), cirrosis hepática y litiasis biliar. Resultados. Se evaluaron 100 pacientes [edad mediana: 26 años (rango: 16-65)]. Mediana de edad al diagnóstico: 2 años. Media IMC 21,5 ± 2,6 DS kg/m2. IP 69/100 (69%), DRFQ 29 pacientes (29%), PA 4 pacientes (4%). Cirrosis hepática 6 pacientes (6%) y litiasis biliar 21 pacientes (21%). Mediana VEF1 50%. Mutación más frecuente ΔF508 (54%). Catorce pacientes (14%) desnutridos. La Pseudomona aeruginosa fue el microorganismo colonizador más frecuente (67%). Se reportó una mediana de sobrevida global de 160 meses (IC 95% 131-160). Se halló menor sobrevida en aquellos pacientes que presentaban IP [(media 124,2 vs. 136 meses (p = 0,4)], desnutrición [mediana 60 vs. 160 meses (p = 0,001)] y cirrosis [mediana 36 vs. 160 meses (p = 0,003)]. Conclusión. Las enfermedades digestivas y la desnutrición influyen negativamente en la sobrevida de los pacientes adultos con FQ.
Palabras claves. Fibrosis quística, páncreas, hígado, adultos, sobrevida.
Impact of Hepatobiliopancreatic Affectation Nutritional Status on the Survival of Adults with Cystic Fibrosis
Summary
Introduction. The increase survival of adult patients with Cystic Fibrosis (CF) has generated a greater relevance of extra pulmonary disease. Objective. Determine digestive, nutritional commitment and its impact on the survival of adult patients with CF. Methods. Retrospective, descriptive and comparative study of 100 adult patients with CF, assisted between 2003 and 2017. It was evaluated: clinical parameters, genetic, body mass index (BMI), respiratory function tests, bacteriological sputum, pancreatic insufficiency (PI), CF-related diabetes (CFRD), acute pancreatitis (AP), liver cirrhosis and biliary lithiasis. Results. 100 patients were evaluated [median age: 26 years (range: 16-65)]. Median age at diagnosis: 2 years. Mean BMI 21.5 ± 2.6 SD kg/m2. PI 69/100 (69%), CFRD 29 patients (29%), AP 4 patients (4%). Liver cirrhosis 6 patients (6%) and biliary lithiasis 21 patients (21%).Median Forced Expiratory Volume in first second (FEV1) 50%. Most frequent mutation ΔF508 (54%). Fourteen patients (14%) malnourished. Pseudomonas aeruginosa was the most frequent colonizing microorganism (67%), with a median overall survival of 160 months (95% CI 131-160). We found lower survival in those patients who had PI [(mean 124.2 vs. 136 months (p = 0.4)], malnutrition [median 60 vs. 160 months (p = 0.001)] and cirrhosis [median 36 vs. 160 months (p = 0.003).] Conclusion. Digestive diseases and malnutrition influence negatively in the survival of adult patients with CF.
Keywords. Cystic fibrosis, pancreas, liver, adults, survival.
Abreviaturas
FQ: Fibrosis quística.
IMC: Índice de masa corporal.
IP: Insuficiencia pancreática.
DRFQ: Diabetes relacionada con FQ.
PA: Pancreatitis aguda.
VEF1: Volumen espiratorio forzado en el primer segundo.
CFTR: Regulador de la conductancia transmembrana de fibrosis quística.
EHAFQ: Enfermedad hepática asociada a la fibrosis quística.
TAC: Tomografía axial computada.
USG: Ultrasonografía.
RMI: Imágenes por resonancia magnética.
CVF: Capacidad vital forzada.
ADA: Asociación Americana de Diabetes.
CFF: Cystic Fibrosis Foundation.
Introducción
La fibrosis quística (FQ) es la enfermedad multisistémica hereditaria autosómica recesiva más frecuente en la raza blanca. Se caracteriza por la presencia de mutaciones en un gen llamado CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), afecta pulmones, páncreas, hígado y otros órganos, y promueve a una disfunción de las glándulas exocrinas, ocasionando secreciones anormalmente espesas.1, 2
La incidencia mundial es de aproximadamente uno de cada 2500-3000 recién nacidos,3 y en nuestro país se estima en uno de cada 6100, según datos del programa de pesquisa neonatal.4
Aproximadamente el 9,9% de los casos se diagnostica recién en la edad adulta.5 En estos pacientes el compromiso pulmonar inicial es más leve y generalmente tienen un páncreas exocrino suficiente, a diferencia de aquellos en los que se diagnostica precozmente.
En los últimos años se halló un notable incremento en la sobrevida de la enfermedad, que dio lugar a una población creciente de adultos que viven con FQ como una enfermedad crónica. En los Estados Unidos, ha llegado a representar el 53% de toda la población afectada por esta enfermedad en el año 2016.6
La FQ presenta grandes variaciones fenotípicas, siendo sus manifestaciones más prevalentes la enfermedad pulmonar y la insuficiencia pancreática exócrina. Entre las manifestaciones gastrointestinales, se destacan las del tubo digestivo (íleo meconial, obstrucción intestinal),7, 8 vías biliares e hígado (colelitiasis, cirrosis biliar)9, 11 y páncreas (pancreatitis aguda, insuficiencia pancreática exocrina y endócrina).11, 14
La enfermedad hepática asociada a la fibrosis quística (EHAFQ) constituye la causa de muerte no pulmonar más importante, representando entre 2,5 a 3,3% de la mortalidad.6, 15 La afectación hepática puede manifestarse desde una colestasis (neonatal), enfermedad del tracto biliar, a cambios histológicos tales como esteatosis, cirrosis y complicaciones como hipertensión portal.16 Aproximadamente un 20-25% de los pacientes con FQ desarrollarán enfermedad hepática, pero solo el 3-7% de estos progresarán a cirrosis.17, 18 Su patogenia comprende un complejo proceso de inflamación, fibrosis, remodelación, apoptosis y colestasis, como resultado de un mal funcionamiento de la proteína CFTR, de reacciones inmunológicas y estrés oxidativo.19, 20 El resultado sería la obstrucción de los pequeños conductos biliares, que conduce a una cirrosis biliar focal, pudiendo progresar en algunos casos a cirrosis multilobular.16
Existen algunos factores de riesgo asociados a EHAFQ, incluyendo el sexo masculino, la IP,10, 15 el antecedente de íleo meconial21 o la mutación severa.22
El objetivo del presente trabajo es determinar el compromiso digestivo (cirrosis hepática, afectación pancreática y biliar), la valoración nutricional y evaluar la sobrevida en pacientes adultos con FQ asistidos en la Unidad de Fibrosis Quística del Hospital Interzonal General de Agudos R. Rossi de la ciudad de La Plata (Argentina).
Métodos
Se realizó un estudio retrospectivo, descriptivo y comparativo de pacientes adultos (≥ 16 años) con diagnóstico de FQ, asistidos en la unidad de FQ del Hospital Rodolfo Rossi de la ciudad de La Plata, desde enero de 2003 hasta junio de 2017. El diagnóstico de FQ se basó en criterios clínicos, test del sudor y/o detección de dos mutaciones del gen CFTR.
Se analizaron las características genéticas, clínicas, antropométricas, bacteriológicas, imágenes ecográficas y pruebas de función respiratoria de cada paciente. Se estableció la concentración de elastasa fecal < 200 µgr/g para diagnóstico de insuficiencia pancreática (IP).
El estudio genético incluyó la búsqueda de mutaciones mediante la metodología PCR ASO reversa; inno-lipa CFTR 19 y 17, innogenetics.
El diagnóstico de pancreatitis aguda (PA) fue establecido por la presencia de dos de tres de los siguientes criterios: dolor abdominal consistente con la enfermedad, amilasa sérica elevada más de tres veces del límite superior normal e imágenes fuertemente sugestivas (tomografía axial computarizada (TAC) contrastada, ultrasonografía (USG) o resonancia magnética (RMI).23 El diagnóstico de diabetes relacionada con FQ (DRFQ) se estableció con glucemia en ayunas superior a 126 mg/dl y a las 2 h superior a 200 mg/dl con una prueba de tolerancia a la glucosa oral con 75 gramos.
El diagnóstico de cirrosis subyacente fue realizado por el hallazgo de cambios histológicos compatibles en tejido hepático, por las características macroscópicas del hígado en el acto operatorio o por las evidencias clínicas y paraclínicas de la enfermedad.24
Se consideró como diagnóstico de litiasis biliar la presencia de material sólido, cálculo o barro, en el tracto biliar y vesícula biliar.25 Los estudios ecográficos se realizaron con un ecógrafo Phillips HD7.
El estatus nutricional se evaluó utilizando el índice de masa corporal (IMC) con la formula peso/altura2 (kg/m2). Según su estado nutricional, los pacientes se dividieron en tres grupos: desnutrición: IMC < 19 kg/m2, en riesgo nutricional: IMC 19-22 kg/m2 para las mujeres y 19-23 kg/m2 para los varones, y óptimo: IMC > 22 kg/m2 para las mujeres y > 23 kg/m2 para los varones.26 La función respiratoria fue evaluada por el volumen espiratorio forzado en el primer segundo (VEF1) y capacidad vital forzada (CVF).
Se confeccionó una planilla de recolección de datos con el software Microsoft Excel 2010. Estos fueron procesados mediante el programa EPI Info versión 7 y MedCalc versión 18,9. Los resultados fueron agrupados en tablas para facilitar su interpretación y los valores se expresan en media ± 1 DS o mediana y rango. Para la comparación de variables cualitativas se utilizó el test de Chi2 o Fisher y T de Student para las cuantitativas. Se consideró diferencia significativa a una p < 0,05. Las estimaciones de la supervivencia se efectuaron mediante el método de Kaplan-Meier. Para comparar curvas de supervivencia se empleó el test logrank.
Resultados
Se incluyeron un total de 100 pacientes adultos (≥ 16 años) con diagnóstico de FQ asistidos por un equipo interdisciplinario de un centro de adultos de la ciudad de La Plata, Argentina. De ellos, 43 mujeres (43%) y 57 varones (57%), con una edad mediana de 26 años (rango: 16-65). La mediana de edad al diagnóstico fue de 2 años (rango: 4 meses-26 años). En 18 pacientes (18%) el diagnóstico de FQ fue realizado en la edad adulta.
Las características referidas al estado pancreático muestran que 69 pacientes (69%) presentaban IP, 4 pacientes (4%) PA y 29 pacientes (29%) DRFQ.
Se halló IP en 6/18 (33,3%) de los pacientes diagnosticados en la edad adulta y en 63/82 (76,8%) de aquellos diagnosticados en la infancia, hallándose una diferencia significativa (p = 0,00007).
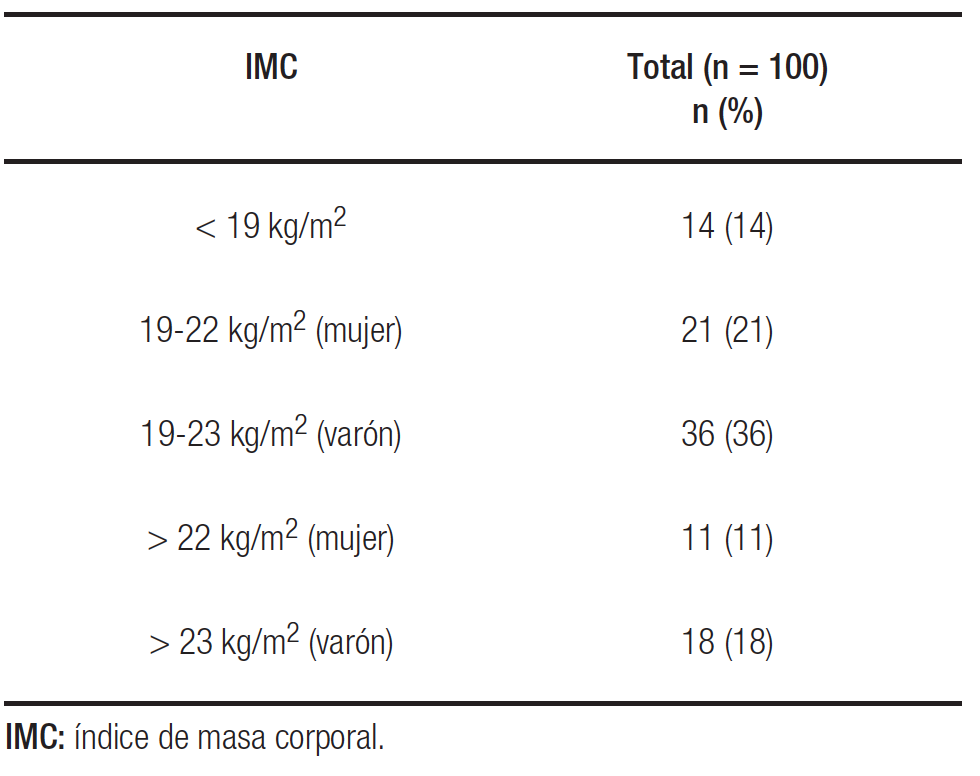
La media del IMC fue 21,5 ± 2,6 kg/m2. El 14% del total de la serie presentaba un IMC por debajo de 19 kg/m2, mientras que 29 pacientes (29%) alcanzaron las metas del IMC presentando niveles óptimos (Tabla 1).
Tabla 1. Distribución según IMC de la totalidad de los pacientes diagnosticados de fibrosis quística incluidos en el estudio
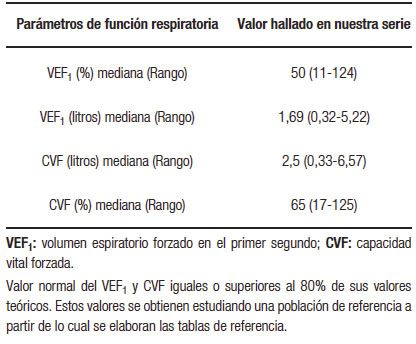
La mediana del VEF1 fue 50 % (rango: 11-124). En la Tabla 2 se describe mediana y rango de los principales parámetros de función respiratoria hallados en la totalidad de los pacientes estudiados.
Tabla 2. Principales parámetros de función respiratoria en la totalidad de los pacientes con FQ
La cirrosis hepática fue diagnosticada en 6 pacientes (6%) de nuestra serie. Todos con diagnóstico asociado de insuficiencia pancreática. Entre los 6 pacientes con cirrosis, solo 1 presentó antecedente de íleo meconial. La media de edad fue de 23,5 ± 4,59 años. La mediana de edad al diagnostico fue de 11 años (rango: 9-15). Solo en 2 pacientes se llegó al diagnostico de cirrosis mediante biopsia hepática. Al compararse la asociación entre los pacientes con antecedentes de íleo meconial y cirrosis, no se halló diferencia significativa (p = 0,4).
Se diagnosticó litiasis biliar a nivel vesicular en 21/100 (21%) (13 varones y 8 mujeres), hallándose una media de edad de 28,5 ± 9,7 (DS) años. Cuatro pacientes estaban colecistectomizados al ingreso a la unidad.
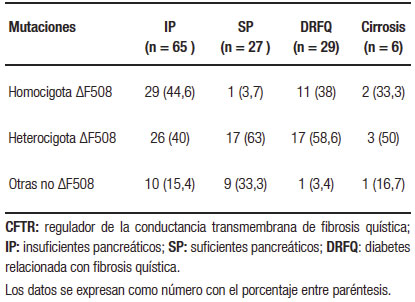
El estudio genético se realizó en 92 pacientes. La mutación más frecuente fue ΔF508 hallada en 100/184 alelos estudiados (54%). Las mutaciones del gen CFTR agrupadas en los pacientes con IP, suficiencia pancreática (SP), DRFQ y cirrosis hepática se detallan en la Tabla 3.
Tabla 3. Mutaciones del gen CFTR distribuidos entre IP, SP, DRFQ y cirrosis, detectadas en los 92 pacientes adultos con fibrosis quística
Entre los microorganismos colonizadores hallados, la Pseudomona aeruginosa fue la más frecuente (67%), siguiendo el Staphylococcus aureus sensible a meticilina (34%), el Staphylococcus aureus resistente a meticilina (30%) y la Burkholderiacepacia (19%). En 45/100 (45%) se observó la asociación de dos o más gérmenes.
Estimación de sobrevida
Se reportó una media de sobrevida de 128,6 meses (IC 95% 113,5-143,7) y una mediana de 160 meses (IC 95% 131-160) (Figura 1).
Figura 1. Distribución según probabilidad de sobrevida global en meses en la totalidad de los pacientes adultos con FQ
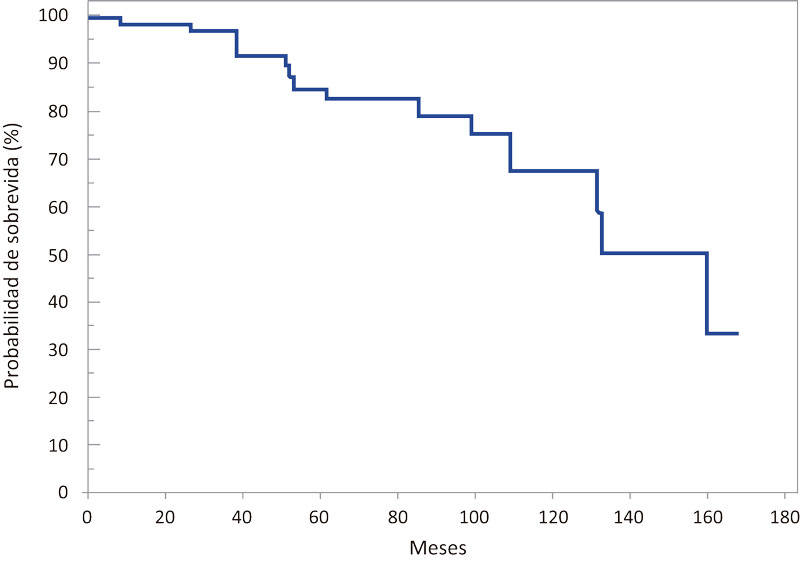
La sobrevida global estimada según el método de Kaplan-Meier fue de 83% a 5 años y 68% a los 10 años. Para el final del período de estudio (30 de junio de 2017), aproximadamente el 67% del total de los pacientes permanecían vivos. La media de sobrevida de los pacientes con insuficiencia pancreática fue de 124,2 meses (IC 95% 107,2-141,1), mientras que en los suficientes pancreáticos se halló una media de sobrevida de 136 meses (IC 95% 107,5-164,4), no hallándose diferencia significativa (p = 0,4) (Figura 2).
Figura 2. Caracterización de la supervivencia según suficientes (SP) e insuficientes pancreáticos (IP) en la totalidad de la muestra de los pacientes con FQ
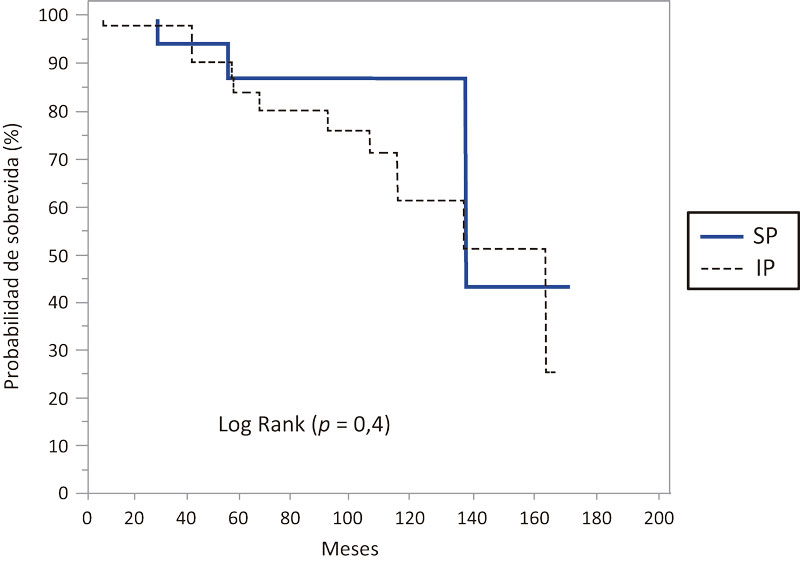
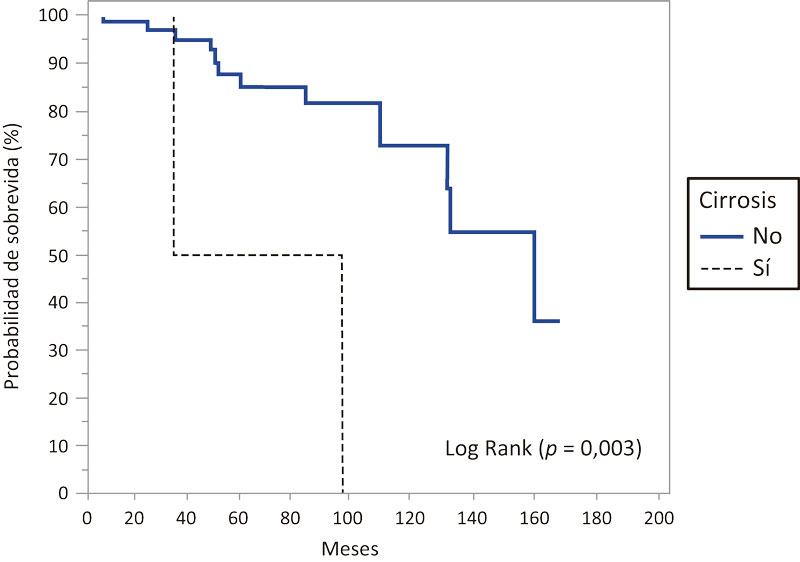
Los pacientes con cirrosis hepática presentaron una media de sobrevida de 67 meses (IC 95% 29,7-104,2), mediana de 36 meses (IC 95% 36-98), mientras que en aquellos que no presentaban cirrosis hepática, la media de sobrevida hallada fue de 133,4 meses (IC 95% 118,3-148,5), mediana de 160 meses (IC 95% 131-160), hallándose una diferencia significativa (p = 0,003) (Figura 3).
Figura 3. Probabilidad de sobrevida en meses en los pacientes estudiados con FQ según se trate de cirróticos y no cirróticos
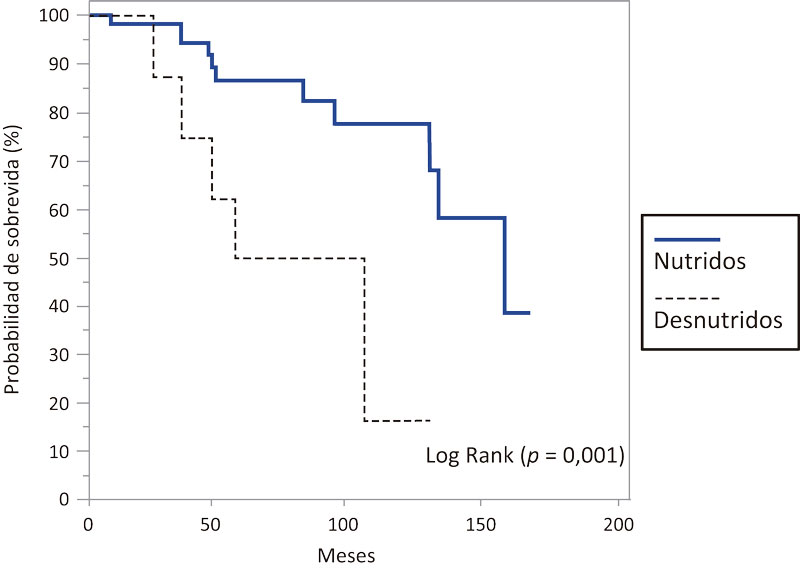
Se halló una media de sobrevida mayor en los pacientes que presentaban un estado de nutrición en riesgo/óptimo (IMC ≥19 kg/m2) que resultó en 135,9 meses (IC95% 120,3-151,5), mediana de 160 meses (IC95%131-160), con respecto a aquellos que presentaban parámetros de desnutrición (IMC < 19 kg/m2) (media de 78,9 meses (IC95% 52,1-105,6), mediana de 60 meses (IC95% 36-108), hallándose diferencia significativa (p = 0,001) (Figura 4).
Figura 4. Probabilidad de sobrevida en meses en los pacientes estudiados con FQ según parámetros de nutrición y desnutrición
Discusión
Si bien la fibrosis quística del páncreas es reconocida como una entidad vinculada estrechamente a la pediatría y a la neumonología, ya en los primeros reportes del pediatra Guido Fanconi en 193627 y, luego, de Dorothy Andersen en 1938,28 se reconoció la asociación entre la fibrosis y la formación de quistes dentro del páncreas con las bronquiectasias, abscesos pulmonares e insuficiencia pulmonar crónica y progresiva.
Del sistema digestivo, el páncreas es el órgano más afectado en la FQ y las células que producen las secreciones exocrinas (páncreas exocrino) son las más frecuentemente dañadas. La función endocrina también puede alterarse. Además puede desarrollarse el daño parenquimatoso inflamatorio como pancreatitis.29 En nuestro centro, en relación con el cuadro clínico y aspectos genéticos relacionados con la afectación pancreática hallamos una prevalencia de IP del 69%, que resulta inferior a la publicada (85%-90%).4 Se halló una mayor prevalencia de IP entre los pacientes diagnosticados de FQ en la infancia (76,8%) en comparación con los diagnosticados en la edad adulta (33,3%), diferencia estadísticamente significativa (p = 0,00007). La presencia de pacientes diagnosticados recién en la edad adulta y con preservación de la función pancreática exocrina podría explicar la menor prevalencia encontrada de IP, en relación con la publicada en otros estudios, la mayor parte de los cuales se llevaron a cabo principalmente en centros pediátricos.
Acerca de la correlación entre genotipo y fenotipo en nuestra serie de pacientes adultos, el genotipo predominante entre los pacientes con IP fue homocigota para la mutación ΔF508, en oposición con los pacientes suficientes pancreáticos en los que predominó el patrón heterocigota u otras mutaciones no ΔF508, diferencia estadísticamente significativa (p = 0,0003).
En concordancia con nuestros resultados, se observa que en estudios de correlación genotipo-fenotipo,30, 31 se ha establecido que los pacientes homocigotos o heterocigotos compuestos por dos mutaciones severas (clases I, II o III) como la mutación ΔF508 y G542X manifiestan insuficiencia pancreática, mientras que otras mutaciones como R334W y R117H (clases IV o V) en uno de sus alelos determinan un fenotipo pancreático suficiente.
La pancreatitis es una complicación poco frecuente en la población con FQ. Ocurre en el 20% de los pacientes suficientes pancreáticos.32 Se ha comunicado también en pacientes insuficientes pancreáticos.33, 34 A pesar de que en nuestra serie, el diagnóstico de FQ fue realizado con anterioridad al episodio de pancreatitis, resulta importante destacar que autores como Foruny Olcina35 señalan que la misma puede ser la manifestación inicial y única de la enfermedad. En el presente trabajo, la prevalencia de pancreatitis fue 4%, superior a la descripta en las tres series más importantes publicadas en la literatura (1,76%,1,84% y 1,76%),33, 34, 36 y similar a la hallada por Sojo Aguirre (3,3%),12 presentándose solamente en los pacientes suficientes pancreáticos. Aunque es muy difícil establecer comparaciones entre las mencionadas series, por las diferencias tan evidentes en la población de pacientes (De Boeck,33 10071 pacientes con FQ publicados vs. 100 pacientes de nuestra serie), consideramos que dado que la pancreatitis se observa preferentemente en la adolescencia o en el adulto joven,34 la mayor prevalencia encontrada en nuestra serie puede deberse a que nuestro centro es de atención exclusiva de adultos.
La diabetes ha evolucionado como una complicación también debido al aumento de la longevidad de los pacientes con FQ. Se asocia con una mayor morbilidad y mortalidad, por lo tanto, el diagnóstico rápido y el manejo agresivo son importantes.37 La Guía de la Asociación Americana de Diabetes (ADA) 2018 clasifica a la DRFQ en la categoría 4, Diabetes por otras causas.38
En lo que respecta a la histopatología en el páncreas de los adultos con FQ, existe una fibrosis progresiva que lleva a atrofia acinar asociada a una infiltración progresiva de células grasas. Se ha encontrado que el número de células beta están reducidas en los pacientes con FQ tanto con diabetes como sin ella,39 y su disfunción es uno de los principales factores fisiopatogénicos en el desarrollo de DRFQ.37
La DRFQ puede aparecer a cualquier edad, siendo la prevalencia de un 9% a los 5-9 años, un 26% a los 10-20 años y aproximadamente hasta un 50% a los 30 años.14, 40 A pesar de que en la mayoría de los centros de atención de adultos, la DRFQ constituye una comorbilidad muy frecuente, la prevalencia en nuestra población fue inferior a la reportada por Moran y col.14 (29% vs. 40-50%).
Como ya se dijo anteriormente, la DRFQ se manifiesta más comúnmente en la adolescencia y la adultez, hallándose un leve predominio en el sexo femenino, en relación con la mutación ΔF508 y en pacientes con insuficiencia pancreática.4 Estos factores que predisponen DRFQ son coincidentes con las características epidemiológicas y clínicas de los 29 pacientes de nuestro centro. El genotipo predominante entre los pacientes con DRFQ fue heterocigota para la mutación ΔF508, hallazgo que se contrapone con lo publicado por otros autores donde predominó el genotipo homocigota ΔF508.41
La función pulmonar y el estado nutricional están estrechamente ligados y ambos son fuertes predictores de morbilidad y mortalidad en pacientes con FQ.42, 43 La desnutrición en FQ es el resultado de un desajuste entre las necesidades energéticas y la ingesta calórica, agravada por malabsorción secundaria a insuficiencia pancreática exocrina crónica.
La European Cystic Fibrosis Society44 establece que en pacientes mayores de 18 años, el valor adecuado del IMC debe mantenerse en 20 kg/m2, aunque idealmente y de acuerdo con el Consenso de la Cystic Fibrosis Foundation (CFF) de los Estados Unidos,6 debe alcanzar 22 kg/m2 en mujeres y 23 kg/m2 en varones. El porcentaje de personas con FQ en los Estados Unidos que alcanzaron estas metas fue incrementándose progresivamente (40% en 2006, 46% en 2011 y 51% en 2016).6 En nuestro estudio, solo el 29% de los pacientes mayores de 20 años registrados hasta 2017 alcanzaron dicha meta, porcentaje sensiblemente inferior al publicado en el Registro de la Fundación Americana de Fibrosis Quística de 2016 (51%)6 y por Barni (43,8%).45
En nuestro centro el IMC promedio fue 21,5 ±2,6 kg/m2, resultado similar al publicado por Barni45 en una serie de 73 pacientes en Brasil que dan una cifra promedio de 21,0 ± 3,0 kg/m2 y una serie de 58 pacientes en Chile publicado por Fernández,46 con un valor de IMC promedio de 22 ± 3,4 kg/m2.
La expresión de CFTR en la vesícula biliar es uno de los más altos de todos los tejidos humanos.47 Por lo tanto, al ser el árbol biliar un sitio importante para la actividad de CFTR, su ausencia puede resultar en enfermedad clínica como resultado de disminución de la secreción de bicarbonato biliar, susceptibilidad a toxinas provenientes de la mucosa intestinal y la interrupción de la circulación enterohepática.16 Dentro de las manifestaciones gastrointestinales en FQ, la litiasis biliar es frecuente, y ocurre en entre el 12% y 24% de los pacientes, dependiendo del criterio de selección.48, 49 En la mayoría de los casos se manifiesta en forma asintomática,50 y se detecta mediante la evaluación ecográfica abdominal anual como método de screening según recomendación del Consenso Nacional de FQ.4 La ecografía tiene una sensibilidad y especificidad mayor al 95% en la identificación de litiasis vesicular.51 Con respecto a la terapéutica, se sugiere la indicación de ácido ursodesoxicólico (20-30 mg/ kg/día).4 Stern y col.49 sugieren la realización de colecistectomía en pacientes con FQ y colelitiasis sintomática, siempre y cuando la función pulmonar sea adecuada. La colecistectomía puede ocurrir en hasta 4% de los pacientes.52 Los resultados obtenidos en nuestra serie de pacientes con respecto a la prevalencia de patología biliar (21%) fueron coincidentes con aquellos descriptos en la bibliografía.
La EHAFQ fue descripta por Andersen28 en 1938. Es una afectación relativamente habitual y aparece como una complicación temprana. Constituye la tercera causa de muerte en los pacientes con FQ,53 aunque los signos y síntomas más elocuentes respiratorios y pancreáticos dejan en segundo plano la importancia que presenta la afectación hepática de la enfermedad. Se manifiesta en la mayoría de los casos con una discreta elevación de las pruebas bioquímicas hepáticas, mientras que solo una pequeña proporción (3-7%) desarrolla una cirrosis hepática. En nuestra serie, los 6 pacientes con cirrosis hepática ingresaron a la unidad de adultos con diagnóstico ya establecido en hospital pediátrico de referencia. Estos datos coinciden con lo aportado en la bibliografía, que sugiere que la enfermedad hepática crónica clínicamente evidente se desarrolla antes o durante la pubertad,54 con una rápida disminución después de los 10 años15 y rara presentación después de los 18 años.17
No se halló asociación estadística entre la presencia de íleo meconial y cirrosis (p = 0,4), coincidiendo con algunos autores19, 22 que sostienen que el rol jugado por este factor en el desarrollo de enfermedad hepática resulta controvertido. Otro factor de riesgo para el desarrollo de cirrosis descripto por algunos autores15 es el género masculino, lo que no fue confirmado en nuestro estudio, ya que se halló en la mitad de los pacientes afectados.
La cirrosis ocurre predominantemente en aquellos individuos con mutaciones severas e insuficiencia pancreática,10, 15 sin existir una correlación genotipo-fenotipo específica, como se observa en la enfermedad pancreática.53 En nuestro estudio de los 6 pacientes con cirrosis, 2 eran homocigotos para la mutación más frecuente ΔF508, 3 eran heterocigotos y el restante presentaba otras mutaciones.
En la actualidad es creciente el número de pacientes adultos debido a una mayor sobrevida en respuesta a una mejor eficacia de los procedimientos diagnósticos y terapéuticos. Asimismo, otro factor importante que contribuyó a mejorar la sobrevida de los pacientes con FQ fue la atención desde la etapa pediátrica en centros especializados asistidos por equipos interdisciplinarios, y es aconsejable continuar también con esta modalidad en la edad adulta.
En los Estados Unidos, según el reporte de la Fundación de Fibrosis Quística,6 la mediana de edad de sobrevida prevista para los nacidos en 2016 fue de 47,7 años, observándose año a año mejoras sustanciales en la sobrevida de las personas con FQ. De acuerdo a datos del Registro de Fibrosis Quística del Reino Unido, Keogh y col.,55 sobre 10428 pacientes estudiados durante el período 2011-2015, concluyen que la mitad de los recién nacidos y adultos de 30 años o más en la actualidad alcanzarían una sobrevida hasta los 50 años. En la Argentina no se hallaron publicados datos epidemiológicos de sobrevida en pacientes con FQ hasta el año 1991, cuando Macri y col.56 publicaron un estudio latinoamericano multicéntrico que incluye a un total de 743 pacientes con diagnóstico de FQ, de los cuales 414 fueron asistidos en un centro pediátrico de la Argentina. Sobre un seguimiento a 21 años, se halló que el 39,2% sobrevivió a los 10 años y el 18% a los 15 años después del diagnóstico. Otro estudio publicado por Lentini y col.57 en el año 2014, sobre 106 pacientes divididos en 4 cohortes, halló que en el año 2008 la sobrevida a los 17 años fue del 80%. En nuestro centro exclusivo de adultos, se halló una sobrevida global de 83% a los 5 años y 68% a los 10 años.
Si bien cada vez es más abundante la bibliografía científica sobre FQ en adultos, la creación de unidades exclusivas de atención de adultos es creciente pero reciente en la Argentina, lo que ocasiona que existan pocas publicaciones que evalúen sobrevida en adultos en nuestra región, y resulte difícil establecer una comparación con nuestros resultados.
En nuestro estudio se observó que los pacientes que presentaban IP mostraron una sobrevida inferior frente a los suficientes pancreáticos (124,2 meses vs. 136 meses), aunque la diferencia entre ambos grupos no resultó estadísticamente significativa (p = 0,4) (Figura 2). En relación con los factores asociados a la sobrevida Kerem y col.58 hallaron en su estudio de 673 pacientes con FQ que si bien el predictor más significativo fue el VEF1 , la sobrevida puede estar influenciada también por la función pancreática, dado que la IP se asocia a maldigestión, malabsorción y desnutrición. Por ello, la prescripción de una terapia de sustitución enzimática pancreática en pacientes con IP ajustada en base al peso del individuo y/o el contenido de lípidos de la dieta es esencial para lograr, alcanzar y mantener un buen estado nutricional.59
En cuanto al estado nutricional, nuestro estudio mostró la influencia negativa de la desnutrición en la sobrevida de los pacientes con FQ (mediana de 160 meses en los pacientes bien nutridos vs. mediana de 60 meses en aquellos que presentaban parámetros de desnutrición) (p = 0,001) (Figura 4), en concordancia con lo ampliamente publicado en la bibliografía.43, 50, 60
Con la mejora en la supervivencia, las manifestaciones hepáticas de la enfermedad emergen como un problema médico cada vez más significativo. Por lo tanto resulta importante su diagnóstico temprano a los efectos de evitar la aparición de complicaciones.61 En nuestro estudio, al compararse la sobrevida entre los pacientes cirróticos y aquellos que no presentaban cirrosis, se halló una diferencia significativa (mediana 36 meses vs. mediana de 160 meses, respectivamente) (p = 0,003) (Figura 3).
En conclusión, las personas con FQ en la Argentina, en consonancia con la tendencia mundial, han prolongado su sobrevida y cada año más pacientes alcanzan la vida adulta. Las enfermedades digestivas junto con la desnutrición influyen negativamente en la sobrevida de los pacientes con FQ, por consiguiente es aconsejable su diagnóstico y terapéutica oportuna.
Aviso de derechos de autor
 © 2021 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
© 2021 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: D´Ascenzo MV, Bosia JD, Borzi SM y col. Impacto de la afectación hepatobiliopancreática y el estatus nutricional en la sobrevida de adultos con fibrosis quística. Acta Gastroenterol Latinoam. 2020;50(1):9-19. https://doi.org/10.52787/vipw2810
Referencias
- Gregory R, Cheng S, Rich D, Marshall J, Paul S, Hehir K, Ostedgaard L, Klinger K, Welsh M, Smith A. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 1990; 347: 382-386.
- Riordan J, Rommens J, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066-1073.
- Rowe S, Miller S, Sorscher E. Cystic fibrosis. N Engl J Med 2005; 352: 1992-2001.
- Castaños C, Rentería F. Consenso Nacional de Fibrosis Quística. Arch Argent Pediatr 2008; 106: 01-52.
- Nick J, Rodman D. Manifestations of cystic fibrosis diagnosed in adulthood. Curr Opin Pulm Med 2005; 11: 513-518.
- Cystic Fibrosis Foundation Patient Registry 2016. Annual data report [Internet] [cited 2017 sept 21]. Disponible en: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2016-Patient-Registry-Reports/.
- Hanly J, Fitzgerald M. Meconium Ileus equivalent in older patients with cystic fibrosis. BMJ 1983; 286: 1411- 1413.
- Heine C, Parada M, Gil R, López F, Lizana C, Fernández M, Quera R. Síndrome de obstrucción intestinal distal en pacientes adultos con fibrosis quística. Casos clínicos Rev med Chile 2010; 138: 68-72.
- Wilschanski M, Durie P. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut 2007; 56(8): 1153-1163.
- Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med 2007; 13(6): 529-536.
- Kelly T, Buxbaum J. Gastrointestinal Manifestations of Cystic Fibrosis. Dig Dis Sci 2015; 60: 1903-1913.
- Sojo Aguirre A, Martínez Ezquerra N, Bousono García C, García Novo M, Heredia González S, López-Manzanares J, Baranda García F, Vázquez Cordero C. Pancreatitis en la fibrosis quística: correlación con el genotipo y estado pancreático. An Pediatr (Barc). 2011; 75: 401-408.
- Brennan A, Beynon J. Clinical updates in cystic fibrosis-related diabetes. Semin Respir Crit Care Med 2015; 36: 236-250.
- Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care 2009; 32: 1626-1631.
- Colombo C, Battezzati P, Crosignani A, Morabito A, Costantini D, Padoan R, Giunta A. Liver disease in cystic fibrosis: a prospective study on incidence, risk factors, and outcome. Hepatology 2002; 36: 1374-1382.
- Debray D, Kelly D, Houwen R, Strandvik B, Colombo C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011; 10 (Suppl 2): S29-S36.
- Nash K, Allison M, McKeon D, Lomas D, Haworth C, Bilton D, Alexander G. A single centre experience of liver disease in adults with cystic fibrosis 1995-2006. J Cystic Fibros 2008; 7: 252-257.
- Lamireau T, Monnereau S, Martin S, Marcotte J, Winnock M, Álvarez F. Epidemiology of liver disease in cystic fibrosis: A longitudinal study. J Hepatol 2004; 41: 920-925.
- Corbett K, Kelleher S, Rowland M, Daly L, Drumm B, Canny G, Greally P, Hayes R, Rourke B. Cystic fibrosis-associated liver disease: A population-based study. J Pediatr 2004; 145: 327-332.
- Więcek S, Woś H, Grzybowska-Chlebowczyk U. Aetiopathogenesis of liver changes in the course of cystic fibrosis, considering disturbances of the bile acid profile as well as genetic and immunological factors. Prz Gastroenterol 2013; 8: 290-294.
- Maurage C, Lenaerts C, Weber A, Brochu P, Yousef I, Roy C. Meconium ileus and its equivalent as a risk factor for the development of cirrhosis: an autopsy study in cystic fibrosis. J Pediatr Gastroenterol Nutr 1989; 9: 17-20.
- Wilschanski M, Rivlin J, Cohen S, Augarten A, Blau H, Aviram M, Bentur L, Springer C, Vila Y, Branski D, Kerem B, Kerem E. Clinical and genetic risk factors for cystic fibrosis related liver disease. Pediatrics 1999; 103: 52-57.
- Banks P, Bollen T, Dervenis C, Gooszen H, Johnson C, Sarr M, Tsiotos G, Vege S. Classification of acute pancreatitis-2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62: 102-111.
- Oberti F, Valsesia E, Pilette C, Rousselet M, Bedossa P, Aubé C, Gallois Y, Rifflet H, Maiga M, Penneau-Fontbonne D, Cales P. Noninvasive diagnosis of hepatic fibrosis or cirrosis. Gastroenterology 1997; 113: 1609-1616.
- Horton J, Bilhartz L. Litiasis biliar y sus complicaciones. En: Feldman M, Friedman L, Sleisenger M. Eds. Sleisenger&Fordtran. Enfermedades gastrointestinales y hepáticas. Fisiopatología, diagnóstico y tratamiento, 7ª ed. Editorial Médica Panamericana 2004: 1126-1152.
- Milla C. Nutrition and Lung Disease in Cystic Fibrosis. Clin Chest Med 2007; 28: 319-330.
- Fanconi G, Kehlinger E, Knauer C. Das Zöliakie-Syndrom bei angeborener cystischer Pankreasfibrose and Bronchiektasien. Wien Med Wochenschrift 86: 753-756.
- Andersen D. Cystic fibrosis of the páncreas and its relation to celiac disease: clinical and pathological study. Am J Dis Child 1938; 56: 344-399.
- Ozen Alahdab Y, Duman D. Pancreatic involvement in cystic fibrosis. Minerva Medica 2016; 107: 427-436.
- Zielenski J, Tsui Lap-Chee. Cystic fibrosis: Genotypic and Phenotypic Variations. Rev. Genetics 1995; 29: 777-807.
- Kere M, Corey M, Kerem B, Rommens J, Markiewicz D, Levison H, Tsui LC, Durie P. The relation between genotype and phenotype in cystic fibrosis: analysis of the common mutation (ΔF508). N Engl J Med 1990; 323: 1517-1522.
- Ooi C, Durie P. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros 2012; 11: 355-362.
- De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics 2005; 115: 463-469.
- Gooding E, Bradley J, Pulenston K, Gyi K, Hodson M. Westaby D. Symptomatic pancreatitis in patients with cystic fibrosis. Am J Gastroenterol 2009; 104: 1519-1523.
- Foruny Olcina J, Moreira Vicente V, Maiz Caro L, Carrera Alonso E, Casals T. Pancreatitis aguda recidivante como forma de presentación de fibrosis quística. Gastroenterol Hepatol 2005; 28: 20-22.
- Durno C, Corey M, Zielenski J, Tullis E, Tsui L, Durie P. Genotype and pancreatitis. Gastroenterology 2002; 123: 1857-1864.
- Lang S. Glucose intolerance in cystic fibrosis patients. Paedriatic respiratory reviews. 2001; 2: 253-259.
- American Diabetes Association. Professional Practice Committee: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018; 41 (Suppl 1): S3-S3.
- Lohr M, Goertchem P, Nizze H, Gould N, GBould V, Oberholzer M, Heitz P, Klöppel G. CF associated islet changes may provide a basis for diabetes. Virchows Arch Pathol Anat 1989; 414: 179-185.
- O’Riordan S, Dattani M, Hindmarsh P. Cystic fibrosis-related diabetes in childhood. Horm Res Paediatr 2010; 73: 15-24.
- Street M, Spaggiari C, Ziveri M, Rossi M, Volta C, Viani I, Grzincich G, Sartori C, Zanzucchi M, Raia V, Terzi C, Pisi G, Zanetti E, Boguszewski M, Kamoi T, Bernasconi S. Insulin production and resistance in cystic fibrosis: effect of age, disease activity, and genotype. J Endocrinol Invest 2012; 35: 246-253.
- Borowitz D. The interrelationship of nutrition and pulmonary function in patients with cystic fibrosis. Curr Opin Pulm Med 1996; 2: 457-461.
- Corey M, McLaughlin F, Williams M, Levison H. A comparison of survival, growth, and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol 1988; 41: 583-591.
- Smyth A, Bell S, Bojcin S, Bryon M, Duff A, Flume P, Kashirskaya N, Munck A, Ratjen F, Schwarzenberg S, Sermet-Gaudelus I, Southern K, Taccetti G, Ullrich G, Wolfe S; European Cystic Fibrosis Society. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines J Cyst Fibros 2014; 13 (Suppl 1): S23-S42.
- Barni G, Forte G, Forgiarini L, Lacerda de Oliveira Abrahão C, Dalcin P. Factors associated with malnutrition in adolescent and adult patients with cystic fibrosis. bras. Pneumol 2017; 43(5): 337-343.
- Fernández P, Labarca G. Fibrosis quística en el adulto: experiencia de un centro de referencia nacional. Rev Med Chile 2012; 140: 841-846.
- Boehm K, Collins F. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest 1994; 93: 347-354.
- Zentber-Munro P. Cystic fibrosis: a gastroenterological cornucopia. Gut 1987; 28: 1531-1547.
- Stern R, Rothstein F, Doershuk C. Treatment and prognosis of symptomatic gallbladder disease in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 1986; 5: 35-40.
- Olveira G, Olveira C. Nutrición, fibrosis quística y aparato digestivo. Nutr Hosp 2008; 23 (Suppl 2): 71-86.
- Bennett G, Balthazar E. Ultrasound and CT evaluation of emergent gallbladder pathology. Radiol Clin North Am 2003; 41: 1203-1216.
- Curry M, Hegarty J. The gallbladder and biliary tract in cystic fibrosis. Curr Gastroenterol Rep 2005; 7: 147-153.
- Flass T, Narkewicz M. Cirrhosis and other liver disease in cystic fibrosis. Journal of Cystic Fibrosis 2013; 12: 116-124.
- Diwakar V, Pearson L, Beath S. Liver disease in children with cystic fibrosis. Paediatr Respir Rev 2001; 2: 340-349.
- Keogh R, Szczesniak R, Taylor-Robinson D, Bilton D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. Journal of Cystic Fibrosis 2018; 17: 218-227.
- Macri C, De Gentile A, Manterola A, Lezana Fernández J, Tomezzoli S, Caldeira Reis F, Largo García I. Epidemiology of Cystic Fibrosis in Latin America: Preliminary Communication. Pediatric Pulmonology 1991; 10: 249-253.
- Lentini E, López-Millan A, Lores l, Pesciullesi M, Ituarte L, Viera T. Fibrosis quística: aumento de la sobrevida en un centro especializado a 10 años de seguimiento. Rev Chil Pediatr 2014; 85: 281-287.
- Kerem E, Reisman J, Corey M, Canny G, Levison H. Prediction of Mortality in Patients with Cystic Fibrosis N Engl J Med 1992; 326: 1187-1191.
- Garriga M, Horrisberger A, Ruiz de las Heras A, Catalán N, Fernández G, Suárez M, Porras N, Monje L. Guía de Práctica Clínica para el manejo nutricional de personas con Fibrosis Quística (GPC-FQ). Rev Esp Nutr Hum Diet 2017; 21: 74-97.
- Stephenson A, Tom M, Berthiaume Y, Singer L, Aaron S, Whitmore G, Stanojevic S. A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur Respir J. 2015; 45: 670-679.
- Parisi G, Di Dio G, Franzonello Ch, Gennaro A, Rotolo N, Lionetti E, Leonardi S. Liver Disease in Cystic Fibrosis: an Update. Hepat Mon 2013; 13: e11215.
Correspondencia: José Daniel Bosia
Calle 158 Nº 610, Berisso (C.P. 1923). Argentina
Tel.: 54 221 4612553 / Cel: 221154091506
Correo electrónico: bosia@intramed.net
Acta Gastroenterol Latinoam 2020;50(1):9-19
