Fernando Sarmiento Quintero,1 Verónica Botero,2 Daniel D´Agostino,3 Laura Delgado Carbajal,4 María Rita Dewaele Olivera,5 Celina Guzmán,6 Edgar Játiva,7 Graciela Teresita Martin,8 Milton Mejía Castro,9 Lourdes Ortiz Paranza,10 María Cecilia Pabón Uego,11 Luis Peña Quintana,12 Rubén E Quirós Tejeira,13 Nelson Ramírez Rodríguez,14 Margarita Ramonet,15 Juan Rivera Medina,16 Marta Sanabria,17 Claudia P Sánchez Franco,18 Lidia Patricia Valdiviezo19
1 Universidad Nacional de Colombia – Fundación HOMI. Bogotá, Colombia.
2 Fundación Valle de Lilí. Cali, Colombia.
3 Hospital Italiano de Buenos Aires. Ciudad Autónoma de Buenos Aires, Argentina.
4 Centro Hospitalario Pereira Rossell-Endoscopía Digestiva. Montevideo, Uruguay.
5 Hospital Pereira Rossell. Montevideo, Uruguay.
6 Universidad Iberoamericana, Hospital La Católica. San José, Costa Rica.
7 Universidad Central del Ecuador. Quito, Ecuador.
8 Hospital Dr A L Castelán de Resistencia Chaco. Corrientes, Argentina.
9 Hospital infantil de Nicaragua. Managua, Nicaragua.
10 Universidad Nacional de Asunción, Facultad de Ciencias Médicas. Asunción, Paraguay.
11 Hospital San José de Melipilla. Santiago de Chile, Chile.
12 Hospital Universitario Materno-Infantil, Universidad de Las Palmas de Gran Canaria, CIBER OBN. Gran Canaria, España.
13 Children’s Hospital & Medical Center. Liver Team, Omaha. Nebraska, Estados Unidos.
14 Universidad Mayor de San Andrés. La Paz, Bolivia.
15 Universidad de Morón. Provincia de Buenos Aires, Argentina.
16 Universidad Nacional Mayor de San Marcos. Lima, Perú.
17 Hospital de Clínicas Universidad Nacional de Asunción. Asunción, Paraguay.
18 Universidad Nacional de Colombia-Fundación HOMI. Bogotá, Colombia.
19 Universidad Científica del Sur. Hospital Nacional Docente Madre Niño San Bartolomé. Lima, Perú.
20 Hospital de Niños “Ricardo Gutiérrez”, Facultad de Medicina (UBA). Ciudad Autónoma de Buenos Aires, Argentina.
Revisaron y corrigieron: Cristina Galoppo20 y Luis Peña12
Nuestro agradecimiento al doctor Valerio Nobili, quien entregó al texto definitivo, sus opiniones y sugerencias.
Acta Gastroenterol Latinoam 2016;46: 246-264
Recibido: 29/10/2015 / Aprobado: 28/06/2016 / Publicado en www.actagastro.org el 03/10/2016

Resumen
Paralelo a la obesidad, la enfermedad por hígado graso no alcolólico (EHGNA) y su progresión a esteatohepatitis (EHNA), se ha convertido en la enfermedad hepática más frecuente en niños y adolescentes. La cirrosis puede ser el evento final en la evolución de esta enfermedad por lo que es importante establecer el diagnóstico en niños con sobrepeso y obesidad. Objetivos. Reunir en un solo escrito los avances en diagnóstico, estrategias para su orientación clínica y manejo de los niños afectados, y a partir de esta revisión, impulsar proyectos de investigación colaborativos en Latinoamérica. Métodos. La SLAGHNP/LAPSGHAN, con la responsabilidad de un editor/coordinador, quien escogió contenido y junto con lo autores la bibliografía con la mejor evidencia, convocó a los asociados a desarrollar esta revisión y se procedió a editar y unificar los contenidos propuestos. Resultados. Se establece claramente la gravedad de la EHGNA/EHNA, como una nueva enfermedad del hígado en niños, así como la poca información que tenemos de ésta en Latinoamérica, al contrario de EE.UU., donde se ha establecido que es la hepatopatía más prevalente en niños. Se reafirma la utilidad de la química sanguínea e imágenes, y aunque se desconoce cuándo está indicada, se establece que la biopsia de hígado es el patrón de oro. Conclusiones. Es importante, oportuno y pertinente tomar la iniciativa del estudio y seguimiento en Latinoamérica de una patología que por su progresión tan rápida planteará en un futuro a mediano plazo un problema de salud pública para toda el área. Debe iniciarse a la brevedad un registro latinoamericano, con los aportes de cada país, liderado por la SLAGHNP/LAPSGHAN, para establecer la epidemiología, determinar su sospecha desde la clínica y seleccionar los exámenes complementarios adecuados y posibles para el diagnóstico de certeza.
Palabras claves. Niños, adolescentes, hígado graso no alcohólico, esteatohepatitis no alcohólica, síndrome metabólico.
Non-alcoholic fatty liver disease (NAFLD): Review and update. Working group of the Latinamerican Society of Pediatric Gastroenterology, Hepatology and Nutrition (LASPGHAN)
Summary
Along with obesity, non-alcoholic fatty liver disease, and its progression to esteatohepatitis, have become the most frequent liver disease among children and teenagers. Since cirrhosis could be an outcome of this condition, it is urgent to test for it in overweight and obese patients. Objectives. To compile in a single article strategies for clinical coaching and handling of children with this condition. On the basis of this revision, to promote research projects. Methods. SLAGHNP/LAPSGHAN, under responsibility and supervision of an editor/coordinator who chose contents and references with the best evidence, called its associates to perform this revision, after which edition and unification of the contents proposed. Results. The severity of EHGNA/EHNA is established as a new infant liver disease and the lack of information on it in Latin America is noted unlike the United State, where its status as the most prevalent hepatopathy among children has been already established. Usefulness of blood chemistry tests and images is emphasized; although it has not been established when it should be recommended, it is clear liver biopsy is the gold standard for detecting the disease. Conclusions. It is important to start in Latin America research and follow-up programs on this disease, whose rapid progression will be continentwise a public health concern in the near future. A latin american register should be started under leadership of SLAGHNP/LAPSGHAN to establish this liver disease’s epidemiology, establish a clinical diagnosis and choose adequate complimentary tests to confirm diagnosis.
Key words. Children, adolescentes, non alcoholic fatty liver disease, non alcoholic steatohepatitis, metabolic syndrome.
Objetivo y metodología
Este trabajo de revisión obedece al interés de la Junta Directiva de la Sociedad Latinoamericana de Gastroenterología, Hepatología y Nutrición Pediátrica (SLAGHNP/ LAPSGHAN) de impulsar no solo el trabajo interdisciplinario, sino de colaborar con la actualización de sus asociados, con un trabajo en el que intervinieron gastroenterólogos pediatras, nutrólogos, nutricionistas y hepatólogos que trabajaron poniendo su conocimiento, interés, tiempo y dedicación para que esta revisión y puesta al día del conocimiento de una de las más frecuentes y graves complicaciones de la obesidad, cumpla el obejetivo de velar por la salud de nuestros niños en Latinoamérica, al ampliar el conocimiento de los médicos y nutricionistas que están dedicados a su cuidado.
La sociedad convocó a sus asociados a conformar el grupo de trabajo para desarrollar esta revisión y con la responsabilidad de un editor/coordinador, se escogió contenido y, en cojunto con los autores, se seleccionó la bibliografía con la mejor evidencia. Una vez seleccionada, se procedió a distribuir los temas de un contenido previamente elegido, para luego editarlos y unificarlos.
Por último, con estas palabras preliminares, quiero expresar mi reconocimiento a la junta directiva, en especial a su presidente, el Dr Eduardo Hebel, por su fructífera tarea de intercomunicar con esta iniciativa a todo el continente y España. De igual manera a todos y cada uno de los colaboradores, que con su empeño, compromiso y la más alta calidad académica, hicieron posible el feliz término de esta revisión y puesta al día.
Introducción
La enfermedad por hígado graso no alcohólico (EHGNA) es un espectro que abarca desde la esteatosis simple, incluye la esteatohepatitis no alcohólica (EHNA), y llega a la cirrosis y sus posibles complicaciones. Es una entidad cada vez más frecuente, si bien la prevalencia y la incidencia son desconocidas tanto en niños como en adultos en Latinoamérica. Esta realidad debe ser considerada para acordar protocolos tendientes a unificar criterios diagnósticos y terapéuticos. En estas circunstancias, es oportuno y urgente que nuestros países realicen el esfuerzo de actualizar conocimientos que permitan el desarrollo de la investigación.
La epidemia de obesidad que azota al mundo ha puesto al descubierto otro tipo de enfermedades y comorbilidades. De éstas, la más grave es el depósito de grasa en el hígado, que va de la esteatosis o enfermedad de hígado graso no alcohólico (EHGNA), o NAFLD de la sigla en inglés, a la hepatitis grasa llamada esteatohepatitis no alcohólica (EHNA), o NASH de su sigla en inglés, hasta la cirrosis.1
La esteatosis o EHGNA es actualmente considerada como la hepatopatía crónica más frecuente en EE.UU., tanto en niños como en adultos, con una prevalencia e incidencia que van en aumento. El diagnóstico temprano, así como la implementación de estrategias de prevención y tratamiento, son actualmente importantes desafíos pediátricos con el objetivo de llegar con precisión al diagnóstico, combinando los antecedentes familiares, las pruebas del laboratorio y estudios de imágenes.2 El estudio histológico en conjunción con el resto de los parámetros clínicos y bioquímicos establece el diagnóstico. La biopsia hepática es la única que puede distinguir la presencia de esteatosis, determinar la actividad inflamatoria, estadificar la fibrosis y efectuar diagnóstico diferencial con otras entidades. Sin embargo, al ser un procedimiento invasivo y con cierto riesgo, su utilización no puede plantearse como estudio de tamizaje ni de seguimiento, planteando un nuevo desafío.3
La enfermedad aún no está bien documentada en nuestra región, su reconocimiento está basado en general por pequeños estudios, basándose en la clínica y biopsias hepáticas, pero dada la diversidad y los escasos reportes, sus conclusiones son incompletas.
La biopsia hepática es necesaria pero tiene sus limitaciones y riesgos, y el tiempo de su indicación no está lo suficientemente aclarado. Progresivamente se han desarrollado e incorporado nuevos estudios para investigar el grado de esteatosis, inflamación y fibrosis, pero paralelamente la elaboración de una norma o guía será un paso esencial para obtener resultados útiles y llegar a un objetivo común.
Antecedentes
Definición
La EHGNA es considerada por algunos autores como la presentación hepática del síndrome metabólico (SM).4, 5 Si bien es más frecuente en escolares y adolescentes, se han reportado casos en pacientes hasta de 3 años debido al importante aumento de sobrepeso y obesidad, siendo esta condición y la insulinorresistencia el mayor riesgo para desarrollarla.
El término esteatohepatitis no alcohólica fue usado por primera vez en 1980,6 al describirse un patrón que recordaba la hepatitis alcohólica en adultos en ausencia de ingesta de alcohol. Tres años después se reportó por primera vez en niños.7 La EHGNA se define en la biopsia hepática como la infiltración grasa de más del 5% de los hepatocitos, no explicada por otras etiologías como: hepatitis B, C, o hepatitis autoinmune, medicamentos, enfermedades metabólicas, fibrosis quística, ingesta de alcohol y enfermedades genético-metabólicas.4-6
Como ya se mencionó, la histololgía es por el momento el estudio que más se aproxima a la definición de EHGNA y EHNA.
Historia y evolución
La primera referencia de la EHGNA la realizó el patólogo de la Clínica Mayo J Ludwing en 1980, que describió a 20 pacientes adultos con una condición clínico-patológica en la que los hallazgos histológicos correspondían a una hepatitis alcohólica, pero sin el consumo de alcohol y de causa desconocida.6 Estaban afectos de esteatohepatitis con disfunción hepática, depósito graso, hepatitis lobular, necrosis focal, cambios inflamatorios, cuerpos de Mallory, diversos grados de fibrosis y en 3 pacientes cirrosis. Los autores ya vislumbraban su asociación con obesidad y diabetes.
En la infancia la primera referencia se remonta a 1983 por JR Moran, que describe 3 niños obesos con elevación de las aminotrasferasas, dolor abdominal inespecífico y biopsia hepática con criterios de EHNA.7
Historia natural de la enfermedad
En general, se considera que los pacientes con esteatosis simple presentan un relativo curso clínico benigno sin progresión histológica, que puede incluso revertir,8 especialmente con la pérdida de peso, o progresar a EHNA y fibrosis como se ha reportado en algunos estudios.9 La presencia de EHNA se convierte en un factor de riesgo para el desarrollo de fibrosis y cirrosis, con cifras que oscilan entre el 8% al 59% en diferentes series publicadas, así como hepatocarcinoma,8-16 mientras la fibrosis avanzada se asocia con un incremento de la mortalidad, independientemente de los factores de riesgo conocidos.17-19
Por otra parte, existen factores étnico-genéticos y ambientales que parecen influir en la evolución; los pacientes afroamericanos tienen menor prevalencia de EHGNA y la fibrosis es menos severa que en los pacientes caucásicos e hispánicos,20 a pesar de un grado similar de obesidad, esteatosis hepática e insulinorresistencia. La evolución a hepatocarcinoma en niños no tiene antecedentes y solo se ha reportado un caso asociado, sin que pueda afirmarse que hizo parte de la esteatosis o corresponda a comorbilidad.21
La prevención de la EHGNA se basará preferentemente en el control de los factores de riesgo modificables como el sobrepeso y los hábitos de vida no saludables, la prevención del bajo peso al nacimiento y la promoción de la lactancia materna.22
Epidemiología
Los principales factores de riesgo para esteatosis hepática son el sobrepeso y la obesidad.23 El estudio de Pajuelo en Perú presenta una prevalencia de sobrepeso y obesidad en niños menores de 5 años de 6,9%, siendo mayor en las áreas metropolitanas, y en Argentina del 20% de sobrepeso y del 5,4% de obesidad en adolescentes.24 En Colombia la prevalencia de exceso de peso (sobrepeso y obesidad) en niños y niñas de 5 a 17 años es del 17,5%.25
La prevalencia de la EHGNA en adultos se ha calculado entre el 20-30% de la población general en los países del occidente asiático y EE.UU.,26, 27 pero la prevalencia global en niños es desconocida. El mayor acercamiento se hizo en un estudio en San Diego entre 1993 y 2003 sobre 742 autopsias en el que encontraron grasa en el hígado en el 13% de las biopsias, definida como mayor del 5%. La prevalencia de hígado graso entre los 2 y 19 años fue del 9,6%, pero subió al 38% en los niños que tuvieron obesidad.28
Los grupos de riesgo para EHGNA en adultos han sido definidos por un aumento del índice de masa corporal (IMC), el síndrome metabólico y la resistencia a la insulina. En pediatría el principal factor de riesgo lo constituye la obesidad.29-32 En 41 adolescentes, quienes tenían un promedio de 59 de IMC, 83% tenían EHGNA y el 20% cumplía criterios histológicos para EHNA.29 En Latinoamérica las cifras son más bajas posiblemente por diferentes condiciones. En Cuba en un estudio en 44 niños obesos entre los 4-16 años, el 48% tenían hígado graso,33 y en Venezuela los estudios muestran una prevalencia promedio de 65% entre niños con sobrepeso y obesidad.34
Los diferentes grupos étnicos también han mostrado prevalencias variables de hígado graso. Los niños hispanos tienen mayor prevalencia de EHGNA que los niños afroamericanos a pesar de que las tasas de obesidad en las dos poblaciones son similares.31, 32
Patogénesis
Tanto la EHGNA como la EHNA son procesos generalmente asintomáticos, por lo tanto, todo paciente con un IMC ≥ 2DE o ≥ 25 kg/m2 debe ser investigado. La gama de este proceso va de solamente acumulación de grasa en el hígado o esteatosis hepática a la presencia de diferentes grados de inflamación con o sin fibrosis, esteatohepatitis. Una vez que la fibrogénesis se presenta, existe el riesgo de progresión a cirrosis.
La patogénesis de la EHNA no es del todo conocida. Probablemente, la teoría más aceptada es la de los “dos golpes”. Todo comienza con la acumulación de grasa en el hígado (primer golpe), lo que conduce a un estrés oxidativo (segundo golpe) que produce inflamación en el hígado.35 La acumulación de grasa se produce por la resistencia a la insulina que lleva a la acumulación de triglicéridos en el hígado. Su acumulación en el hepatocito ocasiona cambios a nivel de la mitocondria, que lleva a falla funcional y producción de radicales libres, que a su vez producen estrés oxidativo, lo cual lleva a la inducción de una respuesta inflamatoria con el riesgo de fibrogénesis.
Una vez establecida la esteatosis, existe el riesgo potencial de desarrollar inflamación o hepatitis, que no evoluciona a fibrosis en todos los casos.3 Los ácidos grasos libres pueden inducir algunas lipogenasas microsomales de los citocromos p-450 que llevan a la producción de radicales libres que son hepatotóxicos y conducen a la inducción de inflamación.35
El patrón inflamatorio de la EHNA en niños es diferente al de los adultos.36 El proceso inflamatorio y de fibrosis inicial en los adultos ocurre en la zona 3 o perisinusoidal, y en los niños, aunque se produce también en la zona 3, es más intenso y más frecuente en la zona 1 o periportal. En forma especulativa esta diferencia estaría explicada por un posible factor hormonal en el momento en que la acumulación de grasa tiene lugar en relación a la pubertad.37
El riesgo de desarrollar esteatohepatitis también parece estar influido por el proceso de inflamación sistémica que ocurre en los niños obesos, por la elevación de la leptina y disminución de los niveles de adiponectina.38
Subsecuentemente, el proceso inflamatorio crónico conlleva al desarrollo de fibrosis; la inflamación crónica estimula la activación de las células estrelladas y progenitoras hepáticas, activación que conduce a una reacción a nivel del ducto biliar con la posterior instalación de fibrosis periportal39 y su progresión a cirrosis.3
En resumen, la obesidad puede llevar a acumulación de grasa en el hígado en relación a la resistencia a la insulina y otros fenómenos asociados a ella. La acumulación de grasa en los hepatocitos puede causar daño a nivel mitocondrial, lo cual lleva al aumento de los radicales libres en un ambiente con disbalance antioxidante. Estos radicales libres inducen un proceso inflamatorio en el hígado o esteatohepatitis. Finalmente, la inflamación crónica induce fibrogénesis y la potencial progresión a la cirrosis.
Factores de riesgo
Obesidad. La obesidad ejerce un impacto negativo sobre la EHGNA en todos los aspectos y estadios de la en fermedad. La mejor evidencia de esta relación es el efecto benéfico que se logra en su manejo, cuando se pierde peso y se mejora el estilo de vida.35, 40
Resistencia a la insulina. Es considerada como un componente fundamental en el desarrollo del síndrome metabólico, que eventualmente puede terminar en diabetes mellitus tipo 2 (DMT2). Tanto la EHGNA como la EHNA están muy ligadas a la resistencia a la insulina, y la relación entre SM y EHGNA41 está plenamente reconocida, puesto que facilita la acumulación de grasa tanto en tejido adiposo abdominal subcutáneo y tejido adiposo abdominal visceral, como en el hígado.
Género. Más común en niños que en niñas (relación 2.1),42 explicado aparentemente por el efecto protector hepático de los estrógenos, que además facilitan la función de la insulina.43, 44 Así como los estrógenos protegen, los andrógenos ejercen un potencial rol negativo al agravar la EHNA.45
Origen étnico. El riesgo relacionado con el origen étnico se ha investigado en grandes poblaciones multiétnicas. Se considera que la prevalencia de hígado graso no alcohólico es más alta en la población de origen hispano (45%) y es más baja entre los afroamericanos (24%). Los caucásicos muestran una prevalencia intermedia (33%).28 Las diferencias étnicas podrían relacionarse con los diferentes grados de resistencia a la insulina, así como de adiposidad central visceral y el índice de masa corporal equivalente, pero también pueden ser el resultado de la genética, así como los factores socioeconómicos, incluyendo el tipo de dieta, el ejercicio y el hábitat.20, 46
Alimentación. En pacientes obesos con EHGNA, la grasa se acumula en el hígado anormalmente en forma de triglicéridos, que se derivan de la lipólisis del tejido adiposo en un 60%, de la lipogénesis de novo en el 26%, y un 15% de la dieta en forma de quilomicrones.47 En este depósito anormal, el desequilibrio dietético entre los ácidos grasos omega-6 y omega-3 parece ser un factor determinante. La fructosa que se consume cada vez más como aditivo en bebidas comerciales puede alterar el metabolismo de los lípidos. Existe evidencia que prueba que el consumo elevado de fructosa desencadena un incremento de la lipogénesis de novo, dislipidemia, resistencia a la insulina y obesidad con depósito de grasa central;48, 49 este estímulo a la lipogénesis de novo, contribuye en el depósito de grasa en la patogénesis de la EHGNA.
Otros factores. En general se reconoce que tanto los factores genéticos como ambientales contribuyen en la patogénesis de la EHGNA. Recientemente hay alguna evidencia según la cual en la etiopatogenia de la EHNA también participan los cambios en la microbiota intestinal y los antioxidantes. La absorción y también la malabsorción de la fructosa pueden alterar la microbiota con las repercusiones correspondientes a nivel hepático.50
Cuándo y cómo sospechar EHGNA/EHNA
La identificación oportuna de los niños con sobrepeso y obesidad es necesaria para evitar el desarrollo de SM y sus complicaciones. La única forma de orientar el diagnóstico de SM es tener en cuenta los criterios que lo definen: obesidad central o abdominal visceral, hipertrigliceridemia, hipertensión arterial (HTA), niveles bajos de colesterol HDL, hiperglicemia e insulinorresistencia.51-55 Para algunos autores la EHGNA forma parte del SM, como definición, y para otros, éste es un factor de riesgo para desarrollarla. En cualquiera de las dos situaciones, todas las estrategias encaminadas a impedir la aparición de SM son igualmente válidas para disminuir el riesgo de EHGNA.
Se han documentado predictores en el examen físico tan sencillos como la acantosis nigricans, marcador bien conocido de resistencia insulínica que pudiera ser signo orientador de EHGNA en más del 90% de los casos.56, 57 Los biomarcadores circulantes, entre los que se destaca la adiponectina, catapepsina-D y citokeratina-18, han mostrado su utilidad para diferenciar entre inflamación simple del hígado y esteatosis.58, 59
De acuerdo con la EPSGAHN, a todos los niños obesos mayores de 3 años se les debe realizar ultrasonido abdominal y función hepática como herramienta válida para identificar infiltración grasa del hígado.1 Asimismo el ultrasonido ha mostrado ser, en pacientes obesos, una buena herramienta para reconocer a quiénes se debe realizar biopsia hepática y confirmar el diagnóstico.60 Igualmente, la circunferencia de la cadera, masa grasa y grasa visceral, tienen una significativa correlación para detectar EHGNA.60
Acercamiento al diagnóstico
Diagnóstico clínico
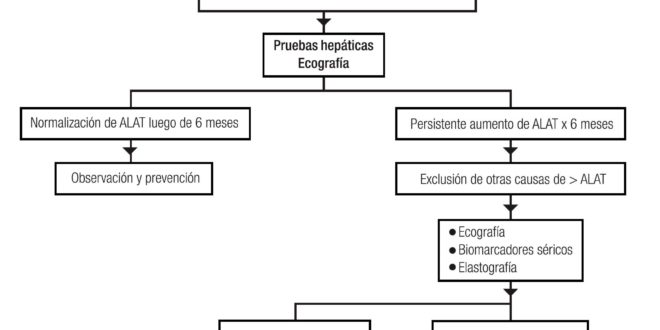
Los numerosos factores que predisponen a la EHGNA dificultan los esfuerzos para diseñar una adecuada orientación para su diagnóstico. La aplicación del algoritmo propuesto en la Figura 1 tiene el objetivo de efectuar un manejo más adecuado.61
El hallazgo casual o no de un incremento de los niveles séricos de alaninoaminotransferasa (ALAT) y/o la presencia de uno de los factores de riesgo (obesidad, sobrepeso, dislipidemia, resistencia a la insulina e hipertensión arterial), debe sugerir la presencia de EHGNA; por lo tanto, se deben solicitar pruebas de función hepática para determinar la presencia de hepatitis: si los niveles de ALAT se normalizan en un período de 6 meses, los niños deben mantenerse bajo observación y realizar tratamiento preventivo (alimentación adecuada y ejercicio); si en cambio, la elevación de ALAT persiste por 6 o más meses, se deben excluir otras causas (hepatitis, ingesta de alcohol, factores genéticos, entre otros) y continuar el estudio con biomarcadores e imágenes. Sin embargo, el hallazgo de una función hepática normal con aminotransferasas normales, no excluye el diagnóstico ya que pueden estar normales o elevarse de forma intermitente, como se ve en la práctica clínica habitual, con el riesgo de diferir el diagnóstico.62 Junto con las pruebas hepáticas se debe hacer una ecografía abdominal sin olvidar que puede quedar sin diagnóstico entre el 5 al 30%.63
En las situaciones que aporten evidencias compatibles con EHGNA, la indicación es dieta hipocalórica y supervisada junto con cambios en el estilo de vida (actividad física controlada), y el potencial uso de medicamentos. En aquellas que no se encuentre una correlación, se requiere la biopsia hepática para confirmar el diagnóstico de EHGNA, antes de comenzar con un tratamiento apropiado (Figura 1).
Figura 1. Algoritmo diagnóstico.
Diagnóstico bioquímico
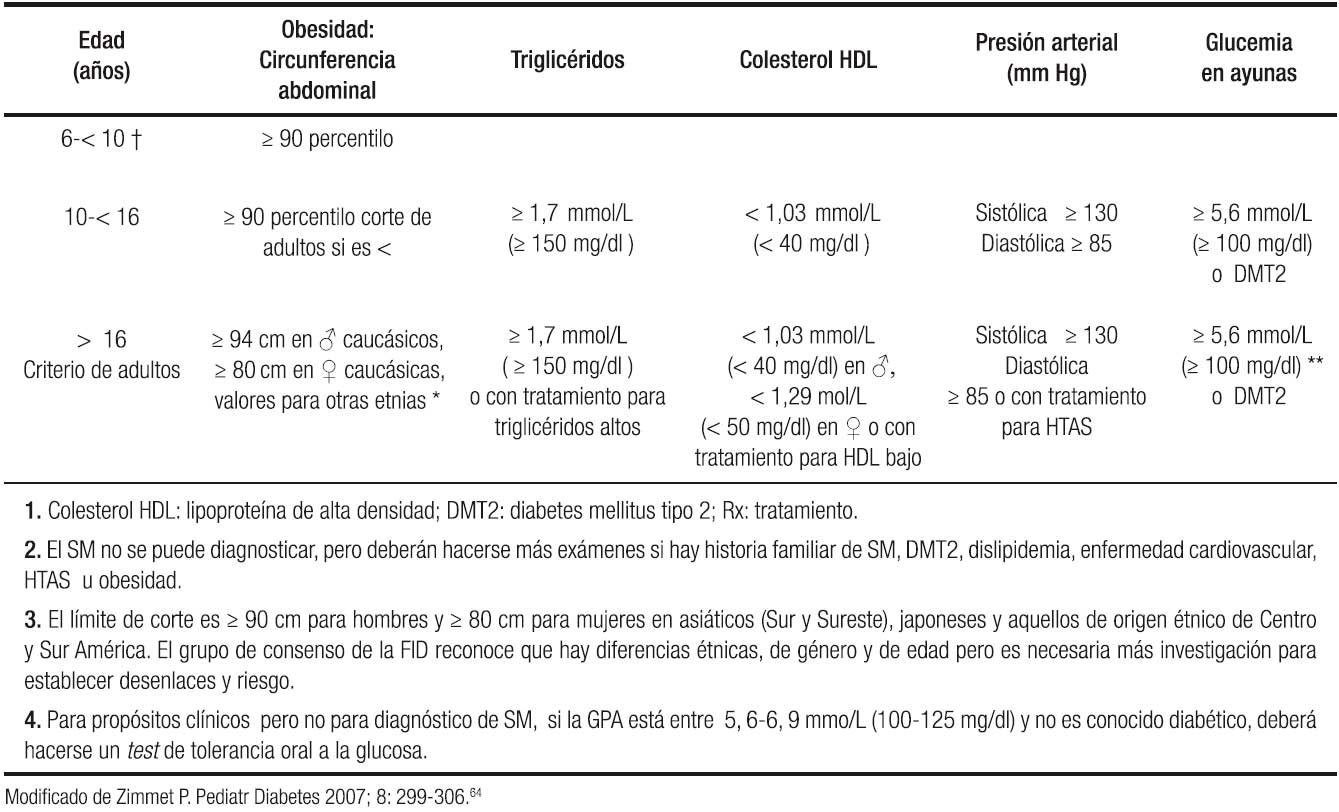
Se deben realizar pruebas bioquímicas en todos los pacientes que tengan sospecha de EHGNA y con mayor fundamento si tienen factores de riesgo reconocidos: género masculino, etnia hispana, edad de transición a la pubertad, sobrepeso y obesidad, alteraciones metabólicas como SM y resistencia a la insulina. De los pacientes con EHGNA, un porcentaje alto tiene criterios de SM, lo que la hace la representación hepática del SM. Entre las definiciones de SM ya señaladas, la FID en el 2007 publicó criterios pediátricos para el SM que se resumen en la Tabla 1 y sirven para orientar en la clínica las pruebas en sangre que se deben solicitar al paciente con criterios de sospecha, junto con la tensión arterial y la CC.64 El diagnóstico del SM requiere la presencia de obesidad central y dos de los otros cuatro factores.
Tabla 1. Orientación para la interpretación bioquímica de la definición de la FID en niños y adolescentes.
Glucemia e insulina: resistencia a la insulina
Varios métodos se han utilizado para determinarlas desde la curva de tolerancia oral a la glucosa, relación glucosa/insulina en ayunas y el índice de sensibilidad a la insulina cuantitativa. El modelo de homeostasis para la resistencia a la insulina (Homeostasis Model Assessment: Insulin Resistence HOMA-IR, por sus siglas en inglés) es el método más certero para cuantificarla,65 y se calcula: HOMA-IR = insulina en ayunas (μU/ml) x glucosa en ayunas (mg/dl) /405.
Se presentan los valores de corte. Prepúberes: niños: 2,67 (sensibilidad 88,2%, especificidad 65,5%), niñas: 2,22 (sensibilidad 100%, especificidad 42,3%). En adolescentes niños: 5,22 (sensibilidad 56%, especificidad 93,3%), niñas: 3,82 (sensibilidad 77,1%, especificidad 71,4%).65 Otros estudios en niños han definido globlalmente el corte de HOMA-IR > 4,39.66
Función hepática
Se recomienda la determinación sérica de enzimas hepáticas, particularmente ALAT, como parte del estudio inicial a estos pacientes.67 La Academia Americana de Pediatría ha recomendado, para el tamizaje de EHGNA en niños con sobrepeso (IMC ≥ 1DE) y obesos (IMC ≥ 2DE), la medición bianual de ALAT.68 Hace algún tiempo, se emitieron estándares, aún vigentes, basados en evidencia para niveles normales de ALAT. En niños: ≤ 25 U/L y niñas: ≤ 22 U/L, según la Encuesta Examinadora Nacional de Salud y Nutrición III, que incluyó sólo niños sin factores de riesgo para enfermedad hepática subyacente.69
Perfil lipídico. La prevalencia del SM y prediabetes aumenta con el aumento de la grasa hepática, y las determinaciones séricas de variables asociadas son fundamentales para la aproximación diagnóstica.65 Los triglicéridos, el colesterol VLDL y el colesterol LDL altos, con un colesterol HDL bajo, son la norma en los sujetos con esteatosis hepática.70, 71
Componentes de autoinmunidad. En algunos casos se debe ser cuidadoso en la interpretación de los anticuerpos autoinmunes, puesto que se ha determinado que en el espectro de EHGNA pueden encontrarse elevaciones discretas de anticuerpos antinucleares (ANA ≥ 1:60), y anticuerpos anti-músculo liso (ASMA ≥ 1:40), que usualmente asociados a niveles normales de IgG, descartan hepatitis autoinmune.72, 73
Otros marcadores en EHGNA. Finalmente se han estudiado una serie de marcadores séricos como la citokeratina-18, que es marcador de apoptosis asociado a esteatohepatitis. Varios estudios han demostrado que la determinación de sus fragmentos puede identificar pacientes con esteatohepatitis con relativa precisión.74, 75
Diagnóstico por imágenes
En el diagnóstico de EHGNA la imagenología es uno de los recursos más utilizados por su seguridad y amplia disponibilidad a pesar del costo elevado a excepción de la ecografía, que además tiene la capacidad de detectar signos de hipertensión portal. Se utilizan 4 diferentes técnicas: ecografía, tomografía computarizada (TC), resonancia magnética (RM) y elastografía.
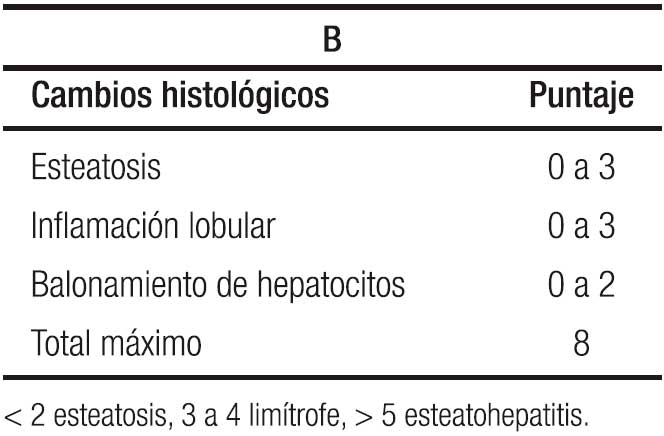
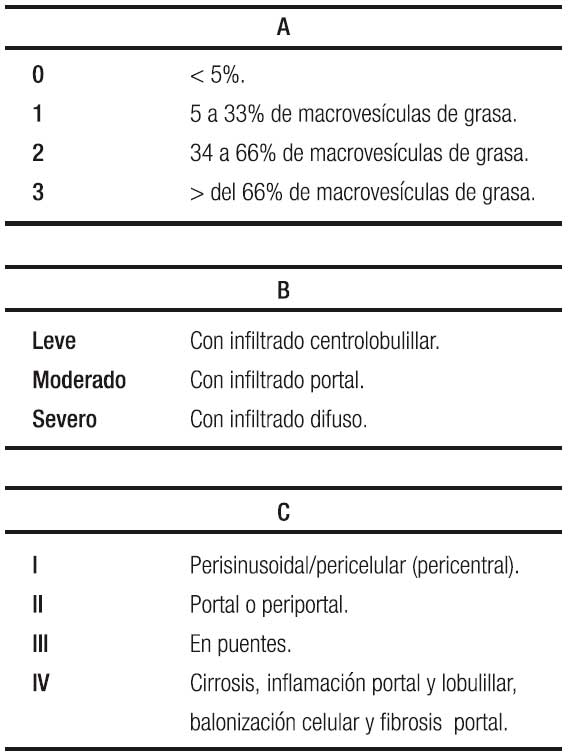
La ecografía hepática se constituye en el examen más utilizado por su nula invasividad y por su buena correlación con los cambios histológicos, con la limitación de no detectar esteatosis por debajo del 30%. Se clasifica de 0 a 3, orden basado en los cambios que se observan de acuerdo al porcentaje de grasa depositada, siendo grado 0 o normal, sin ecorrefringencia hepática; grado 1 o leve, con cambios mínimos de la ecorrefringencia con normal visualización del diafragma y de los bordes de la porta; grado 2 o moderada con cambios moderados de la ecorrefringencia y mínima alteración de la visualización del diafragma y de los bordes de la porta; grado 3 o severa con hiperrefringencia, pérdida de la visualización del diafragma y de los bordes de la porta, con difícil identificación del segmento posterior del lóbulo derecho.76 Para llegar a la comprensión de la ecografía, es necesario repasar la clasificación histológica en la que se contemplan tres ítems: esteatosis, inflamación lobulillar y balonamiento, con puntaje de 0 a 3 los dos primeros y de 0 a 2 para el tercero para un máximo de 8 puntos, Tabla 2A y 2B, y aparte la fibrosis con puntaje de 0 a 4.77 En detalle la esteatosis se define como: 0 ≤ 5%; 1 ≥ 5% a 33%; 2 ≥ 34% a 65%; y 3 ≥ 66%; cuando el paciente tiene esteatosis grados 2 y 3 la ecografía es capaz de detectarla con una sensibilidad y especificidad del 80% y del 86% respectivamente,76 cuando el depósito de grasa en el hígado está por encima del 30%. En resumen, la ecografía o ultrasonografía tiene un rendimiento moderado.78
La TC es una técnica que revela una imagen ecogénica dada por la infiltración de grasa en el hígado, basada en las características de los rayos X de penetrar los tejidos con valores de atenuación denominados unidades de Hounsfield que disminuye la densidad del parénquima y que se comparan con los valores de atenuación del bazo. No ser operador dependiente es su principal característica, pero ocasionar notable exposición a irradiación que es la desventaja.79
La RM ofrece imágenes para la cuantificación de la grasa con una adecuada exactitud sin ninguna invasividad o exposición a radiación, y al contrario de la ecografía, tiene la capacidad de discriminar la esteatosis y la fibrosis. Resulta ser el método imagenológico más certero, con un costo alto que lo limita.80
La elastografía por ultrasonido (FibroScan) y elastografía por resonancia magnética hepática son técnicas de ultrasonografía que miden la velocidad de propagación de ondas de radiofrecuencia a través de un tejido. Específicamente en EHGNA/EHNA, inicialmente se ha empleado el FibroScan para evaluar el grado de fibrosis hepática,81 y el poder diferenciar la fibrosis independiente de la grasa y la inflamación, aumenta su rendimiento. La elastografía por ultrasonido se basa en la disminución de la velocidad de la transmisión de las ondas de radiofrecuencia en la medida en que el hígado aumente su consistencia por la infiltración de grasa; se ve afectada por el aumento de IMC y no puede detectar los grados de esteatosis.82 La elastografía por resonancia magnética se contempla como un método de mayor rendimiento que el FibroScan y no baja su rendimiento con el espesor de la grasa abdominal subcutánea.83 En resumen, estas técnicas novedosas están en experimentación y faltan estudios para determinar su real utilidad en niños.
Diagnóstico histológico
El estudio de la biopsia hepática es la prueba de oro porque identifica con certeza los cambios grasos; además de evaluar la inflamación (esteatohepatitis) y distinguir el grado de fibrosis, determina la evolución de la misma a la cirrosis.84 Incluye la presencia mixta de macro y microvacuolas con el predominio de las primeras, con desplazamiento del núcleo en más del 5% de hepatocitos, balonización, inflamación portal y perisinusoidal, megamitocondrias, cuerpos acidófilos y acumulación de glucógeno en los núcleos, con fibrosis que puede ser porto-portal y porto-centro-lobulillar o perisinusoidal.
Schwimmer caracterizó la esteatosis y la esteatohepatitis, y diferenció el compromiso hepático con el adulto, sugiriendo dos tipos de patrones histológicos: el tipo 1, semejante al adulto con hígado graso mixto, con o sin fibrosis perisinusoidal e inflamación lobular; y el tipo 2 descrito predominantemente en el 51% de los niños con esteatosis macrovesicular con componente inflamatorio portal, sin balonización, con o sin fibrosis.85
El Instituto Nacional de Diabetes, Enfermedades digestivas y Renales patrocinó un Programa de Investigación para desarrollar el puntaje de actividad de esteatosis (EHGNA) denominado NAS (Tablas 2A, 2B y 3).77, 86

Indicación de la biopsia
La edad del paciente es fundamental; cuando el paciente tiene un diagnóstico ecográfico de hígado graso, o alteraciones en el hepatograma con alteración enzimática, se la debe considerar en los siguientes escenarios:87
- Menores de 3 años: luego de descartar exhaustivamente enfermedades genéticas, metabólicas y sindromáticas.
- Entre 3 a 10 años: si hay obesidad y se descartan enfermedades virales, tóxicas, autoinmunes, metabólicas (enfermedad de Wilson).
- En mayores de 10 años: si la ecografía y el hepatograma no se han modificado luego de 6 meses de manejo con cambios en el estilo de vida (alimentación y actividad física), la obesidad es central visceral o hay antecedentes familiares de síndrome metabólico.
La histología que no debe ser propuesta como tamizaje y se debe acompañar de: cociente aminotransferasas/ plaquetas, concentración en sangre de vitamina D, fibrotest y FibroScan. En todos los casos se deben descartar enfermedades tratables, tener un seguimiento ordenado secuencial, dar manejo médico integral por lo menos por 6 meses y considerar pronóstico y riesgo, antes de ordenar la biopsia. En las Tablas 4A, 4B y 4C se detalla la clasificación de Brunt.85, 86, 88
Diagnóstico diferencial
La resistencia a la insulina es un hallazgo común en EHGNA y en consecuencia varias de las características clínicas asociadas tales como la obesidad, la DMT2 y dislipidemia son comorbilidades frecuentes en los niños que la padecen.2, 89
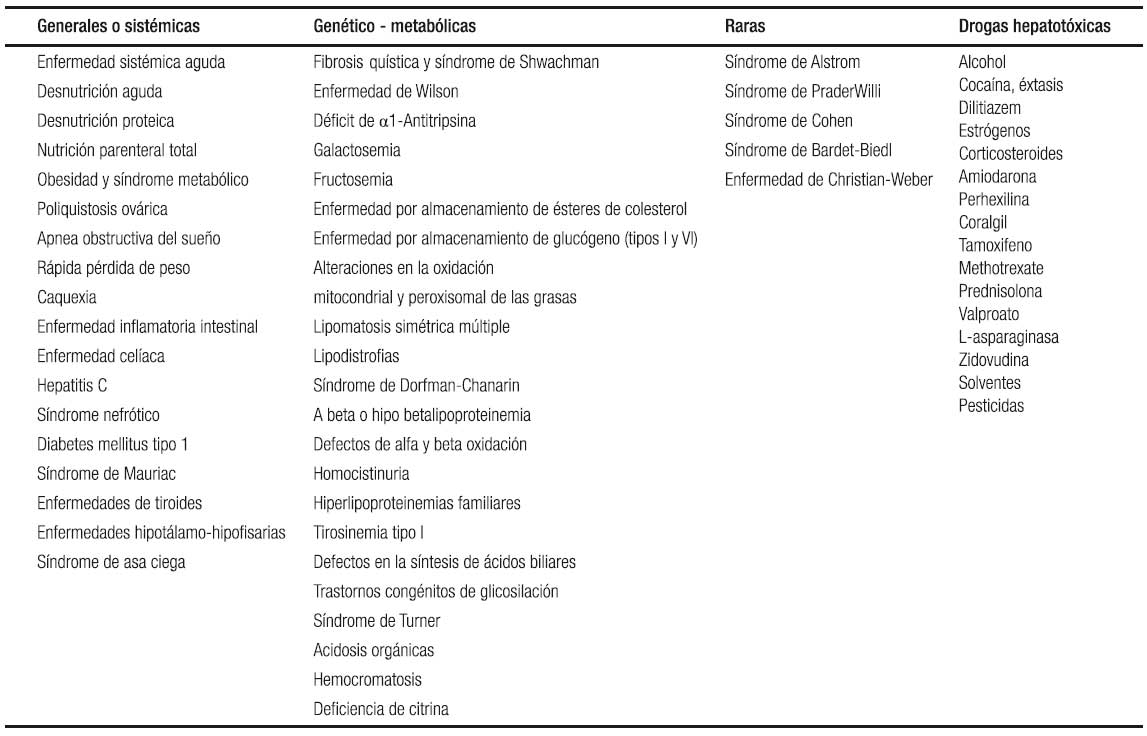
En todos los casos se deben descartar en el diagnóstico diferencial otras etiologías que incluyen enfermedades nutricionales, infecciosas, metabólicas y tóxico-medicamentosas, que puedan cursar con esteatosis hepática. Las pruebas de laboratorio incluyen: serología para las hepatitis virales A, B y C, HIV, niveles de ceruloplasmina, acil-carnitina, test del sudor, α-1antitripsina y estudio genético (genotipo), auto-anticuerpos antinucleares, anti-músculo liso, IgG, anti-transglutaminasa IgA, anti-microsomales hepáticos y renales, anti-mitocondriales, y estudios para errores congénitos del metabolismo (lactato/piruvato en orina, ácidos orgánicos y aminoácidos en suero) y deficiencia de lipasa ácida.90 En la Tabla 5 se enumeran las causas de hígado graso en niños.
Manejo integral
Manejo general: intervención sobre los factores de riesgo, cambios en el estilo de vida.
Los factores de riesgo para el desarrollo de hígado graso no alcohólico en niños y adolescentes se pueden dividir en:91
- No modificables: constitucionales, sexo masculino, etnia hispánica92 y genéticos.
- Modificables: constitucionales (obesidad visceral, resistencia a la insulina, deficiencia de estrógenos) y dietéticos (alta ingesta de fructosa, sacarosa y ácidos grasos omega 6 y baja ingesta de ácidos grasos omega 3).93
Los factores predictivos de evolución de esteatosis a cirrosis son: circunferencia de la cadera, hiperglicemia, hipertensión arterial, hiperinsulinemia en ayunas y el aumento de las enzimas hepáticas.94 Las intervenciones sobre los factores de riesgo y cambios en el estilo de vida se resumen en la Tabla 6.
El descenso de peso tiene un efecto favorable sobre la mejoría significativa en los biomarcadores de estrés oxidativo y el patrón de omega 3, asociado a la capacidad de su biosíntesis que tiene un rol central en el tratamiento de la obesidad y alteraciones metabólicas ocasionadas por la esteatosis hepática. Como las comorbilidades (síndrome metabólico, pre-diabetes y DMT2) empeoran la severidad del hígado graso,91 el ejercicio físico es el mayor y más eficiente estímulo para disminuir el depósito de grasa. El ejercicio físico estimula la biogénesis mitocondrial (el tejido muscular metaboliza más del 75% de los azúcares y las grasas), lo que incrementa notablemente la sensibilidad a la insulina,95 con el aumento del número de transportadores de glucosa musculares GLUT 4, disminuyendo la resistencia a la insulina. En un plan de alimentación, la reducción de la ingesta de sacarosa y fructosa podría ejercer un efecto benéfico dado que la fructosa, podría tener un rol como pro-inflamatorio a nivel hepático.96
Tabla 6. Estrategias en el tratamiento de EHGNA.

Manejo farmacológico
Los cambios en el estilo de vida son hasta ahora la terapia más aceptada en EHGNA y comorbilidades asociadas,97 pero en algunos casos se requiere la administración de terapia farmacológica como parte del manejo para impedir la progresión de la enfermedad. Se utiliza como coadyuvante en pacientes que no tengan buena respuesta a las modificaciones en el estilo de vida.98, 99 Entre las ayudas farmacológicas que se han ofrecido se destacan:
- Vitamina E y D: la eficacia de la vitamina E no es mayor que la que produce la modificación en el estilo de vida y no es superior al placebo en cuanto al cambio de los valores de ALAT, pero se ha demostrado en la histología la mejoría en la balonización hepatocitaria.100, 101 Mientras que para la vitamna E hay evidencia razonable para estar utilizándola desde hace más de una década, aún no se ha establecido si la deficiencia de la D tiene efecto sobre el metabolismo de los lípidos.102, 103
- Metformina: es un agente que aumenta la sensibilidad a la insulina, disminuye la producción de glucosa en hígado y aumenta la utilización de la misma en el músculo; pero al igual que la vitamina E, no tiene mayor eficacia que los cambios dietéticos y el estilo de vida.104
- Ácido ursodeoxicólico: podría ser un hepatoprotector, al evitar el daño mitocondrial que producen las sales biliares activando vías antiapoptóticas y al favorecer las funciones inmunomoduladoras. Su uso en niño con EHGNA no ha demostrado beneficio.105
- Probióticos: se conoce de antemano que la alteración del epitelio intestinal, en su función de barrera, aumenta el nivel de toxinas en sangre y éstas a su vez pueden coadyuvar con el daño inflamatorio en la esteatosis; los probióticos, al mejorar la función de barrera e impedir el sobrecrecimiento bacteriano, disminuyen la endotoxemia y podrían ser factor protector en la patogénesis de la EHGNA, mejorando los valores de ALAT, independientemente del IMC.106
- Ácidos grasos poliinsaturados: tienen efectos antiinflamatorios y además aumentan la sensibilidad a la insulina en los tejidos periféricos; a largo plazo (2 años) disminuyen los niveles de triglicéridos y mejoran los valores de ALAT.107
Nuevas terapias
- Inhibidores de la enzima convertidora de angiotensina: la utilidad de los IECAS se basa en la capacidad que tiene el sistema renina-angiotensina sobre la sensibilidad a la insulina, por lo que su bloqueo podría, como se ha demostrado en algunos ensayos, disminuir los niveles de ALAT y la lesión necroinflamatoria en las biopsias.108
- Las combinaciones de vitamina E más DHA y de estas dos con Colina son estrategias en curso. En estudios separados a la fecha, están pendientes sus resultados.98
Se debe tener en cuenta que, con el incremento de la obesidad en niños, la EGHNA potencialmente puede llegar a EHNA, por lo que se debe estar alerta para hacer prevención.
El futuro en el manejo del hígado graso
Modificación de la microbiota intestinal – Probióticos
En los últimos años numerosos investigadores han sugerido la modulación de la microbiota intestinal por probióticos, prebióticos y simbióticos como un posible enfoque para la obesidad y el hígado graso no alcohólico.106
Existen numerosos reportes, tanto en niños con sobrepeso como con obesidad, que demuestran que el intestino presenta una alteración en la microbiota intestinal109, 110 o disbiosis, que explica por qué el papel de la microbiota intestinal en la EHGNA ha ganado considerable atención.111 Como ya se señaló en la EHGNA hay disrrupción de la barrera intestinal y producción de diversas sustancias pro-inflamatorias con actividad directa a nivel hepático, situación que se conoce como teoría eje hígado-intestino.112 Además, la microbiota intestinal podría estar fuertemente influida por el consumo de grasas y azúcares refinados, lo que estimularía al sistema inmunológico innato, iniciando la cascada inflamatoria.106 Son numerosos los trabajos que confirman los efectos benéficos de los probióticos.111, 113 Se ha demostrado que los niños obesos con EHGNA tratados con Lactobacillus GG evidenciaron una significativa disminución (hasta normalización en el 80% de los casos) en los valores séricos de ALAT,113 además del colesterol total, lipoproteína de alta densidad, TNF-α, con mejoría de la sensibilidad a la insulina.112
El efecto benéfico de los probióticos se demuestra cada vez con mayor fuerza, y el VSL#3, multicepas y especies, es el más estudiado en EHGNA.114 Su administración por 4 meses redujo el contenido graso del hígado evaluado por ecografía, así como una disminución significativa del IMC.105 Además del efecto sobre la grasa hepática, se ha reportado disminución de sustancias pro-inflamatorias, TNF-α, interleuquina-6 y lipopolisacáridos.115
En resumen, la restauración de la microbiota intestinal podría resultar en la normalización de la permeabilidad intestinal, aumento en la producción de ácidos grasos de cadena corta (AGCC) y hormonas intestinales anorexígenas (incluyendo GLP-1 y GLP-2),112 así como la mejoría de la sensibilidad a la insulina.106
Pentoxifilina, silimarina y bitartrato de cisteamina
Pentoxifilina: es un derivado de las metilxantinas, con propiedades hemorreológicas y es usado comúnmente en el tratamiento de la claudicación intermitente, mejora la flexibilidad de los glóbulos rojos, disminuye la viscosidad, mejora la glicolisis aeróbica y el consumo de oxígeno en los tejidos isquémicos. Además, en estudios humanos y animales se ha mostrado como un inhibidor no específico de la fosfodiesterasa, dando como resultado varios cambios fisiológicos a nivel celular: incrementa los niveles de AMP cíclico y disminuye el gen transcriptor del TNF-α, afectando varios pasos de la vía de citoquinas-quemoquinas. Como en modelos humanos y animales con EHGNA se encuentran incrementos séricos del TNF-α entre las citoquinas proinflamatorias, la pentoxifilina se ha planteado como un posible tratamiento.116
Silimarina: Es un derivado de la planta Silybum marianum,117 es un antioxidante natural usado en forma general durante siglos para enfermedades hepáticas.118-120 Algunos reportes usando solo silimarina o en combinación con otros agentes en pacientes con EHNA han mostrado beneficios, pero son estudios limitados, la mayoría a reportes de medicina alternativa. Estudios realizados en ratas sugieren estabilización en la membrana mitocondrial e inhibición del estrés oxidativo.118, 121 Sin embargo, faltan estudios para recomendar su uso en humanos.119, 120
Bitartrato de cisteamina: es una pequeña molécula de aminotiol que es fácilmente transportada a través de la membrana celular.122 Se ha usado en cistinosis y actúa reduciendo la acumulación intralisosomal de cisteína; también tiene efecto antiapoptótico y antioxidante.123 En el manejo de la EHGNA, específicamente en niños, la terapia con cisteamina por 24 semanas redujo las aminotransferasas, la adiponectina total, la leptina y fragmentos de citoqueratina, mecanismos invocados para explicar su beneficio.123, 124
Receptores toll-like
Los receptores toll-like (TLR por sus siglas en inglés) son proteínas transmembrana que pertenecen a una de las 4 familias de los denominados receptores de reconocimiento de la inmunidad innata. No solo se encuentran en las membranas celulares de los macrófagos y de las células dendríticas; también están presentes en las membranas de células epiteliales, endoteliales y fibroblastos, que sin ser células especializadas en la inmunidad innata, participan del proceso de respuesta inflamatoria. También algunos TLR se expresan en los linfocitos T.125 Los TLR reconocen epítopes bacterianos, virales o fúngicos, y partículas endógenas de daño celular, participando en los mecanismos de autoinmunidad y en la respuesta injerto contra receptor.126 Los TLR tienen que ver con el daño hepático, al participar en el estímulo que activa las citoquinas proinflamatorias TNF-α, IL-, e IL-6. Además de los linfocitos, la activación del sistema inmune vía TLR dentro de los monocitos juega un papel importante, por ejemplo, en la fisiopatología de la DMT2.127,128 La expresión de los TLR se ha observado en todos los tipos de células hepáticas: hepatocitos,129, 130 células de Kupffer,129-131 células endoteliales sinusoidales,132 células estrelladas hepáticas,133 células epiteliales biliares,129 así como las células inmunes dendríticas del hígado.132
De la misma forma como la síntesis de TNF-α, IL-6 e IL-1, puede ser estimulada por el aumento de glucosa y de ácidos grasos circulantes en distintos tejidos, además del adiposo, incluyendo hígado y músculo; también se activa por el estímulo de los TLR, acciones que sumadas en la EHNA llevan a inflamación y fibrosis progresiva. En las enfermedades crónicas del hígado, incluyendo NASH, la permeabilidad intestinal resulta alterada por la disfunción de las uniones estrechas epiteliales, la cual sugiere una exposición permanente a altos niveles de TLR, ligados a productos bacterianos provenientes del intestino que podrían llevar a la lesión hepática.134 Al mismo tiempo, el IL-1β estimula en las células hepáticas estrelladas la producción de fibrosis,135 y los TRL-2 en ausencia de colina participan en la inflamación y fibrosis del hígado, siendo las células de Kupffer las que expresan el TRL-2.136
Por otro lado, para explicar el daño hepático cuando el paciente ingiere dietas ricas en grasas, se ha observado en pacientes con EHNA con altos niveles de triglicéridos en sangre, la presencia de ácidos palmítico y esteárico ligados a TLR-4.137 En particular, el ácido laúrico se ha encontrado en el inicio de la señalización del TLR-4 a nivel de la línea de los macrófagos.138 A pesar de lo demostrado, es claro que se requiere mayor investigación.
Receptores de ácidos biliares (Farnesoid X receptor)
El receptor nuclear de ácidos biliares (Farnesoid X Receptor) FXR (gen NR1H4) es una proteína que pertenece a la superfamilia de receptores nucleares que son activados por ligandos y se consideran sensores metabólicos.139 El FXR está involucrado principalmente en la regulación de la homeostasis de los ácidos biliares, así como también en el metabolismo de otros lípidos y de la glucosa, funciones preventivas de tumorogénesis hepática e intestinal, regeneración hepática, mantenimiento de la integridad de la barrera intestinal y atenuación de efectos producidos por enfermedades colestásicas, a través de dos vías (principal y secundaria).140 Sus cuatro isoformas se expresan de manera específica en cada tejido; en hígado aparecen predominantemente dos FXRα1 (+) y FXRα1 (-).141 Recientemente han sido publicados estudios en fase 2 de investigación en adultos que apoyan el uso del ácido obeticólico (OCA), conocido también como INT-147 (derivado 6α etílico del ácido quenodeoxicólico CDCA), agonista selectivo de primera clase del receptor nuclear de ácidos biliares (FXR), a una dosis de 25 mg/día durante 72 días.142 Los resultados han mostrado incremento de la sensibilidad a la insulina, regulación de la homeostasis de la glucosa, modulación del metabolismo de los lípidos, efectos anti-inflamatorios, con disminución de la balonización hepatocitaria y anti-fibróticos con consecuente disminución de los marcadores de inflamación y fibrosis hepática en adultos con diabetes y EHGHA/EHNA.143 En niños esta terapia podría ser promisoria, si bien aún está en fase experimental en adultos.
Péptido 1 similar al glucagón
El péptido 1 similar al glucagón (GLP-1) es un péptido secretado por las células L endocrinas del intestino delgado. Forma parte de las hormonas peptídicas gastrointestinales integrantes del grupo de las incretinas. Su primera función conocida es la estimulación de la producción de insulina por las células β del páncreas. Se secreta a la circulación esplénica y portal en respuesta a la ingesta de alimento postprandial. Una vez en la circulación, la GLP-1 tiene una vida media corta (de 1 a 2 minutos) debido a su degradación rápida por la presencia de la enzima dipeptidilpeptidasa 4 (DPP 4).144
Inicialmente ha sido estudiado por sus efectos sobre el metabolismo de la glucosa, al reducir la glucosa plasmática y al mejorar la sensibilidad a la insulina mediante el aumento de su liberación postprandial, disminución de la secreción de glucagón y retraso del vaciamiento gástrico. Efectos adicionales incluyen la reducción de la ingesta de energía, aumento de la saciedad y la pérdida de peso, por lo que se ha usado como tratamiento en la DMT2 y se ensaya en el tratamiento de la obesidad extrema.145 En otros estudios se ha demostrado que existen receptores para GLP-1 en los hepatocitos humanos y se ha planteado su participación en el metabolismo de los lípidos reduciendo la esteatosis hepática.146
En pacientes con DMT2 tratados con análogos del GLP-1 a los cuales se les realizó resonancia magnética, se ha demostrado disminución progresiva del depósito de lípidos en el hígado y disminución de GLP-1 o aumento de DPP 4 en biopsias hepáticas en pacientes con esteatosis hepática.147
Se han experimentado las acciones de los agonistas de los receptores de los GLP-1, como las exenatida y liraglutida, y los inhibidores de la DPP 4 (sitagliptina, sexagliptina y linagliptina), en su papel de disminuir la esteatosis en individuos con infiltración grasa del hígado. Por lo tanto, se necesita mayor conocimiento en cuanto a su real efecto sobre el metabolismo de los ácidos grasos. Es necesario diseñar estudios clínicos en niños que evalúen tanto la mejoría de la esteatosis como la esteatohepatitis y los posibles efectos secundarios para poder recomendar su uso.
Conclusiones
La obesidad en niños y en adultos se ha convertido en una pandemia cuya consecuencia inmediata es el depósito de grasa en el hígado, a tal punto que se ha convertido en la primera causa de hepatopatía tanto en adultos como en niños. Al contrario de Estados Unidos, en Latinoamérica se desconoce el comportamiento epidemiológico de la EHGNA/EHNA y por lo tanto no es posible la iniciación de las acciones correspondientes para contrarrestar un problema de salud pública muy grave y creciente. El conocimiento de su patogenia y los factores que inciden en su desarrollo con su alarmante incremento, son el paso para tomar las primeras decisiones: reconocer el problema y los factores desencadenantes, e implementar los estudios multicéntricos que nos permitan medir la prevalencia e incidencia para iniciar las acciones preventivas de un problema, tan desconocido, como que su desarrollo no solo se da al poner en juego los factores genéticos y los epigenéticos en el curso del crecimiento del niño y su transición a la edad adulta, si no también, su presencia desde la época del desarrollo embriológico, como recientemente se ha descrito.148 Significa, para el colmo de nuestro desconocimiento, que nos estamos enfrentando a una enfermedad que se puede originar desde el comienzo de la vida in útero.149-151
Referencias
- Vajro P, Lenta S, Socha P, Dhawan A, Mc Kiernan P, Baumann U, Durmaz O, Lacaille F, McLin V, Nobili V. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr 2012; 54: 700-713.
- Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology 2009; 50: 1282-1293.
- Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009; 58: 1538-1544.
- Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol 2015; 21: 11053-11076.
- Berardis S, Sokal E. Pediatric non-alcoholic fatty liver disease: an increasing public health issue. Eur J Pediatr 2014; 173: 131-139.
- Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980; 55: 434-438.
- Moran JR, Ghishan FK, Halter SA, Greene HL. Steatohepatitis in obese children: a cause of chronic liver dysfunction. Am J Gastroenterol 1983; 78: 374-377.
- Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, Becker U, Bendtsen F. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut 2004; 53: 750-755.
- Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, Chim AM, Yu J, Sung JJ, Chan HL Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut 2010; 59: 969-974.
- Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413-1419.
- Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 1990; 11: 74-80.
- Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006; 43: S99-S112.
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005; 129: 113-121.
- Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006; 44: 865-873.
- Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011; 34: 274-285.
- Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med 2011; 43: 617-649.
- Kim D, Kim WR, Kim HJ, Therneau TM. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 2013; 57: 1357-1365.
- Rafiq N, Bai C, Fang Y, Srishord M, Mc Cullough A, Gramlich T, Younossi ZM. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol 2009; 7: 234-238.
- Stepanova M, Rafiq N, Makhlouf H, Agrawal R, Kaur I, Younoszai Z, Mc Cullough A, Goodman Z, Younossi ZM. Predictors of all-cause mortality and liver-related mortality in patients with non-alcoholic fatty liver disease (NAFLD). Dig Dis Sci 2013; 58: 3017-3023.
- Santoro N, Feldstein AE, Enoksson E, Pierpont B, Kursawe R, Kim G, Caprio S. The association between hepatic fat content and liver injury in obese children and adolescents: effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care 2013; 36: 1353-1360.
- Nobili V, Alisi A, Grimaldi C, Liccardo D, Francalanci P, Monti L, Castellano A, de Ville de Goyet J. Non-alcoholic fatty liver disease and hepatocellular carcinoma in a 7-year-old obese boy: coincidence or comorbidity? Pediatr Obes 2014; 9: e99-e102
- Giorgio V, Prono F, Graziano F, Nobili V. Pediatric non alcoholic fatty liver disease: old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr 2013; 13:40.
- Alavian SM, Mohammad-Alizadeh AH, Esna-Ashari F, Ardalan G, Hajarizadeh B. Non-alcoholic fatty liver disease prevalence among school-aged children and adolescents in Iran and its asociation with biochemical and anthropometric measures. Liver Int 2009; 29: 159-163.
- Pajuelo J, Cuadros M, Campos M, Sánchez J. Prevalencia de sobrepeso y obesidad en niños menores de cinco años en el Perú 2007-2010. Rev Peru Med Exp Salud Pública 2011; 28: 222-227.
- Encuesta Nacional de la Situación Nutricional en Colombia. En: http://www.dane.gov.co/files/investigaciones/boletines/vehiculos/bol_veh_Itrim11.pdf ICBF 2010. Bogotá.
- Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004; 40: 1387-1395.
- Nomura H, Kashiwagi S, Hayashi J, Kajiyama W, Tani S, Goto M. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn J Med 1988; 27: 142-149.
- Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006; 118: 1388-1393.
- Xanthakos S, Miles L, Bucuvalas J, Daniels S, García V, Inge T. Histologic spectrum of nonalcoholic fatty liver disease in morbidly obese adolescents. Clin Gastroenterol Hepatol 2006; 4: 226-232.
- Roberts E. Pediatric nonalcoholic fatty liver disease (NAFLD): a «growing» problem? J Hepatol 2007; 46: 1133-1142.
- Schwimmer J, McGreal N, Deutsch R, Finegold M, Lavine J. Influence of Gender, Race, and Ethnicity on Suspected Fatty Liver in Obese Adolescents. Pediatrics 2005; 115: e561-e566.
- Schwimmer JB, Deutsch R, Rauch JB, Behling C, Newbury R, Lavine JE. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J Pediatr 2003; 143: 500-505.
- Pacheco L, Piñeiro LR, Fragoso AT, Valdés AMC, Martínez R. Hígado graso no alcohólico en niños obesos. Rev Cubana Pediatr [revista en la Internet] 2006 [citado 2015 Sep; 78. En: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034- 75312006000100002&lng=es].
- Pontiles M, Morón A, Rodríguez H, Perdomo G. Prevalencia de la enfermedad de hígado graso no alcohólico en una población de niños obesos en Valencia, Venezuela. Archivos Latinoamericanos de Nutrición 2014; 64: 73-82. En: http://www.alanrevista.org/ ediciones/2014/2/?i=art1.
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002; 346: 1221-1231.
- Patton HM Lavine JE, Van Natta ML, Schwimmer JB, Kleiner D, Molleston J; Nonalcoholic Steatohepatitis Clinical Research Network. Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008; 135: 1961-1971.
- Patton HM, Sirlin C, Behling C, Middleton M, Schwimmer JB, Lavine JE. Pediatric nonalcoholic fatty liver disease: a critical appraisal of current data and implications for future research. J Pediatr Gastroenterol Nutr 2006; 43: 413-427.
- Quirós-Tejeira RE, Rivera CA, Ziba TT, Mehta N, Smith CW, Butte NF. Risk for nonalcoholic fatty liver disease in Hispanic youth with BMI > or = 95th percentile. J Pediatr Gastroenterol Nutr 2007; 44: 228-236.
- Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, Weltman MD, Tilg H, Moschen AR, Purdie DM, Demetris AJ, Clouston AD. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 2007; 133: 80-90.
- Sahoo K, Sahoo B, Choudhury AK, Sofi NY, Kumar R, Bhadoria AS. Childhood obesity: causes and consequences. J Family Med Prim Care 2015; 4:187-192.
- Seidell JC. Obesity, insulin resistance and diabetes: a worldwide epidemic. Br J Nutr 2000; 83: S5-S8.
- Wiegand S, Keller KM, Röbl M, L’Allemand D, Reinehr T, Widhalm K, Holl RW. APV-Study Group and the German Competence Network Adipositas. Obese boys at increased risk for nonalcoholic liver disease: evaluation of 16.390 overweight or obese children and adolescents. Int J Obes (Lond) 2010; 34: 1468-1474.
- Villa A, Della Torre S, Stell A, Cook J, Brown M, Maggi A. Tetradian oscillation of estrogen receptor is necessary to prevent liver lipid deposition. Proc Natl Acad Sci U S A 2012; 109: 11806-11811.
- Kelly DM, Nettleship JE, Akhtar S, Muraleedharan V, Sellers DJ, Brooke JC, McLaren DS, Channer KS, Jones TH. Testosterone suppresses the expression of regulatory enzymes of fatty acid synthesis and protects against hepatic steatosis in cholesterol-fed androgen deficient mice. Life Sci 2014; 109: 95-103.
- Zhang H, Liu Y, Wang L, Li Z, Zhang H, Wu J, Rahman N, Guo Y, Li D, Li N, Huhtaniemi I, Tsang SY, Gao GF, Li X. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J Lipid Res 2013; 54: 345-357.
- Dongiovanni P, Anstee QM, Valenti L. Genetic predisposition in NAFLD and NASH: impact on severity of liver disease and response to treatment. Curr Pharm Des 2013; 19: 5219-5238.
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005; 115: 1343-1351.
- Kanerva N, Sandboge S, Kaartinen NE, Männistö S, Eriksson JG. Higher fructose intake is inversely associated with risk of nonalcoholic fatty liver disease in older Finnish adults. Am J Clin Nutr 2014; 100: 1133-1138.
- Moore JB, Gunn PJ, Fielding BA. The role of dietary sugars and de novo lipogenesis in non-alcoholic fatty liver disease. Nutrients 2014; 6: 5679-5703.
- Vos MB. Nutrition, nonalcoholic fatty liver disease and the microbiome: recent progress in the field. Curr Opin Lipidol 2014; 25: 61-66.
- Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Geneva, World Health Organization 1999.
- National Cholesterol Education Program, Executive summary of the third report of the National Cholesterol Education Program (NCEP), Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285: 2486-2497.
- Balkau B, Charles MA: Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet Med 1999; 16: 442-443.
- Alberti KG, Zimmet P, Shaw J. IDF Epidemiology Task Force Consensus Group. The metabolic syndrome a new worldwide definition. Lancet 2005; 366: 1059-1062.
- Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, Allen K, Lopes M, Savoye M, Morrison J, Sherwin RS, Caprio S. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 2004; 350: 2362-2374.
- Castro AL, Arriaga HE, Palacios GC. Esteatosis hepática (EH) como factor asociado a la presencia de riesgo metabólico en escolares y adolescentes obesos. Gaceta Médica de México 2014; 150: 95-100.
- Maclaren NK, Gujral S, Ten S, Motagheti R. Childhood obesity and insulin resistance. Cell Biochem Biophys 2007; 48: 73-78.
- Walenbergh SM, Houben T, Hendrikx T, Jeurissen ML, van Gorp PJ, Vreugdenhil AC, Adriaanse MP, Buurman WA, Hofker MH, Mosca A, Lindsey PJ, Alisi A, Liccardo D, Panera N, Koek GH, Nobili V, Shiri-Sverdlov R. Plasma cathepsin d levels: a novel tool to predict pediatric hepatic inflammation. Am J Gastroenterol 2015; 110: 462-470.
- Feldstein AE, Alkhouri N, De Vito R, Alisi A, Lopez R, Nobili V. Serum cytokeratin-18 fragment levels are useful biomarkers for nonalcoholic steatohepatitis in children. Am J Gastroenterol 2013; 108: 1526-1531.
- Monteiro PA, Antunes B de M, Silveira LS, Christofaro DG, Fernandes RA, Freitas Junior IF. Body composition variables as predictors of NAFLD by ultrasound in obese children and adolescents. BMC Pediatr 2014; 14: 25.
- Alisi A, Feldstein AE, Villani A, Raponi M, Nobili V. Pediatric nonalcoholic fatty liver disease: a multidisciplinary approach. Nat Rev Gastroenterol Hepatol 2012; 9: 152-161.
- Peña L, Aguiar Santana I, Ruiz Moreno. Esteatosis hepática y esteatohepatitis En: F Arguelles Martín, MD García Novo, P Pavón Belinchón, E Román Riechmann, G Silva García, A Sojo Aguirre eds. Tratado de Gastroenterología, Hepatología y Nutrición Pediátrica aplicada de la Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica. Madrid: Ergon; 2011; 577- 585. ISBN: 978-84-8473-891-6.
- Sundaram SS, Zeitler P, Nadeau K. The Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease in children. Curr Opin Pediatr 2009; 21: 529-535.
- Zimmet P, Alberti KG, Kaufman F, Tajima N, Silink M, Arslanian S, Wong G, Bennett P, Shaw J, Caprio S; The metabolic syndrome in children and adolescents – an IDF consensus report. IDF Consensus Group. Pediatr Diabetes 2007; 8: 299-306.
- Kurtoğlu S, Hatipoğlu N, Mazıcıoğlu M, Kendirici M, Keskin M, Kondolot M. Insulin resistance in obese children and adolescents: HOMA-IR cut-off levels in the prepubertal and pubertal periods. J Clin Res Pediatr Endocrinol 2010; 2: 100-106.
- Kelly A, Dougherty S, Cucchiara A, Marcus CL, Brooks LJ. Catecholamines, adiponectin, and insulin resistance as measured by HOMA in children with obstructive sleep apnea. Sleep 2010; 33: 1185-1191.
- Barlow SE. Expert Committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007; 120: S164-S192.
- Schwimmer JB, Dunn W, Norman GJ, Pardee PE, Middleton MS, Kerkar N, Sirlin CB. SAFETY study: alanine aminotransferase cut-off values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010; 138: 1357-1364.
- Strauss RS, Barlow SE, Dietz WH. Prevelance of abnormal serum aminotransferase values in overweight and obese adolescents. J Pediatrics 2000; 136: 727-133.
- Cali AM, De Oliveira AM, Kim H, Chen S, Reyes-Mugica M, Escalera S, Dziura J, Taksali SE, Kursawe R, Shaw M, Savoye M, Pierpont B, Constable RT, Caprio S. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology 2009; 49: 1896-1903.
- Cali AM, Zern TL, Taksali SE, de Oliveira AM, Dufour S, Otvos JD, Caprio S. Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect proatherogenic state. Diabetes Care 2007; 30: 3093-3098.
- Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 2005; 42: 132-138.
- Sanyal AJ, Banas C, Sargeant C, Luketic VA, Sterling RK, Stravitz RT, Shiffman ML, Heuman D, Coterrell A, Fisher RA, Contos MJ, Mills AS. Similarities and differences in outcomes of cirrhosis due to nonalcoholic steatohepatitis and hepatitis C. Hepatology 2006; 43: 682-689.
- Wieckowska A, Zein NN, Yerian LM, Wieckowska A, Zein NN, Yerian LM, López AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006; 44: 27-33.
- Feldstein AE, Wieckowska A, López AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 2009; 50: 1072-1078.
- Shannon A, Alkhouri N, Carter-Kent C, Monti L, Devito R, López R, Feldstein AE, Nobili V. Ultrasonographic quantitative estimation of hepatic steatosis in children With NAFLD. J Pediatr Gastroenterol Nutr 2011; 53: 190-195.
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp- Arida A, Yeh M, McCullough AJ, Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41: 1313-1321.
- Csendes GP, Paolinelli GP, Busel MD, Venturelli AV, Rodríguez J. Hígado graso: Ultrasonido y correlación anatomopatológica. Rev Chil Radiol [online] 2004; 10: 50-52.
- Kim DY, Park SH, Lee SS, Kim HJ, Kim SY, Kim MY, Lee Y, Kim TK, Khalili K, Bae MH, Lee JY, Lee SG, Yu ES. Contrast-enhanced computed tomography for the diagnosis of fatty liver: prospective study with same-day biopsy used as the reference standard. Eur Radiol 2010; 20: 359-366.
- Bohte AE, van Werven JR, Bipat S, Stoker J. The diagnostic accuracy of US, CT, MRI and 1H-MRS for the evaluation of hepatic steatosis compared with liver biopsy: a meta-analysis. Eur Radiol 2011; 21: 87-97.
- Nobili V, Vizzutti F, Arena U, Abraldes JG, Marra F, Pietrobattista A, Fruhwirth R, Marcellini M, Pinzani M. Accuracy and reproducibility of transient elastography for the diagnosis of fibrosis in pediatric nonalcoholic steatohepatitis. Hepatology 2008; 48: 442-448.
- Chan WK, Nik Mustapha NR, Mahadeva S. Controlled attenuation parameter for the detection and quantification of hepatic steatosis in nonalcoholic fatty liver disease. J Gastroenterol Hepatol 2014; 29: 1470-1476.
- Huwart L, Sempoux C, Vicaut E, Salameh N, Annet L, Danse E, Peeters F, ter Beek LC, Rahier J, Sinkus R, Horsmans Y, Van Beers BE. Magnetic resonance elastography for the noninvasive staging of liver fibrosis. Gastroenterology 2008; 135: 32-40.
- Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2010; 7: 195-203.
- Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, Lavine JE. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005; 42: 641-649.
- Brunt EM, Kleiner DE, Wilson LA, Unalp A, Behling CE, Lavine JE, Neuschwander-Tetri BA; NASH Clinical Research Network. A list of members of the Nonalcoholic Steatohepatitis Clinical Research Network can be found in the Appendix. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009; 49: 809-820.
- Carter-Kent C, Yerian LM, Brunt EM, Angulo P, Kohli R, Ling SC, Xanthakos SA, Whitington PF, Charatcharoenwitthaya P, Yap J, López R, McCullough AJ, Feldstein AE. Nonalcoholic steatohepatitis in children: a multicenter clinicopathological study. Hepatology 2009; 50: 1113-1120.
- Alkhouri N, De Vito R, Alissi A, López R, Feldstein A, Nobili V. Development and validation of a new histological score for pediatric nonalcoholic fatty liver disease. Journal of Hepatology 2012; 57: 1312-1318.
- Rodríguez G, Gallego S, Breidenassel C, Moreno LA, Gottrand F. Is liver transaminases assessment an appropriate tool for the screening of non-alcoholic fatty liver disease in at risk obese children and adolescents? Nutr Hosp 2010; 25: 712-717.
- Baratta F, Pastori D, Del Ben M, Polimeni L, Labbadia G, Di Santo S, Piemonte F, Tozzi G, Violi F, Angelico F. Reduced Lysosomal Acid Lipase Activity in Adult Patients With Non-alcoholic Fatty Liver Disease. EBioMedicine 2015; 2: 750-754.
- Marzuillo P, Miraglia E, Santoro N. Pediatriac non-alcoholic fatty liver disease: new insights and future directions. World J Hepatol 2014; 6: 217-225.
- Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004; 40: 1387-1395.
- Yasutake K, Nakamuta M, Shima Y, Ohyama A, Masuda K, Haruta N, Fujino T, Aoyagi Y, Fukuizumi K, Yoshimoto T, Takemoto R, Miyahara T, Harada N, Hayata F, Nakashima M, Enjoji M. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: the significance of dietary cholesterol. Scand J Gastroenterol 2009; 44: 471-477.
- Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001; 121: 91-100.
- Díaz Bustos E, Saavedra C, Meza J. Guía contemporánea de Salud y Ejercicio. Conarem, Santiago de Chile 2007.
- Basaranoglu M, Basaranoglu G, Sabuncu T, Sentürk H. Fructose as a key player in the development of fatty liver disease. World J Gastroenterol 2013; 19: 1166-1172.
- Mager DR, Roberts EA. Nonalcoholic fatty liver disease in children. Clin Liver Dis 2006; 10: 109-131, vi-vii.
- Della Corte C, Liccardo D, Ferrari F, Alisi A, Nobili V. Current pharmacotherapy for treating pediatric nonalcoholic fatty liver disease. Expert Opin Pharmacother 2014; 15: 2501-2511.
- Nobili V, Alkhouri N, Alisi A, Della Corte C, Fitzpatrick E, Raponi M, Dhawan A. Nonalcoholic fatty liver disease: a challenge for pediatricians. JAMA Pediatr 2015; 169: 170-176.
- Lavine JE. Vitamin E treatment of nonalcoholic steatohepatitis in children: a pilot study. J Pediatr 2000; 136: 734-738.
- Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, Abrams SH, Scheimann AO, Sanyal AJ, Chalasani N, Tonascia J, Ünalp A, Clark JM, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011; 305: 1659-1668.
- Dasarathy J, Periyalwar P, Allampati S, Bhinder V, Hawkins C, Brandt P, Khiyami A, McCullough AJ, Dasarathy S. Hypovitaminosis D is associated with increased whole body fat mass and greater severity of non-alcoholic fatty liver disease. Liver Int 2014; 34: e118-e127.
- Bril F, Maximos M, Portillo-Sánchez P, Biernacki D, Lomonaco R, Subbarayan S, Correa M, Lo M, Suman A, Cusi K. Relationship of vitamin D with insulin resistance and disease severity in non-alcoholic steatohepatitis. J Hepatol 2015; 62: 405-411.
- Nobili V, Svegliati-Baroni G, Alisi A, Miele L, Valenti L, Vajro P. A 360-degree overview of paediatric NAFLD: recent insights. J Hepatol 2013; 58: 1218-1229.
- Della Corte C, Alisi A, Iorio R, Alterio A, Nobili V. Expert opinion on current therapies for nonalcoholic fatty liver disease. Expert Opin Pharmacother 2011; 12: 1901-1911.
- Alisi A, Bedogni G, Baviera G, Giorgio V, Porro E, Paris C, Giammaria P, Reali L, Anania F, Nobili V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2014; 39: 1276-1285.
- Alisi A, Nobili V. Non-alcoholic fatty liver disease in children now: lifestyle changes and pharmacologic treatments. Nutrition 2012; 28: 722-726.
- Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology 2004; 40: 1222-1225.
- Ly NP, Litonjua A, Gold DR, Celedón JC. Gut microbiota, probiotics, and vitamin D: interrelated exposures influencing allergy, asthma, and obesity? J Allergy Clin Immunol 2011; 127: 1087-1094.
- Million M, Lagier JC, Yahav D, Paul. M. Gut bacterial microbiota and obesity. Clin Microbiol Infect 2013; 19: 305-313.
- Wong VW, Won GL, Chim AM, Chu WC, Yeung DK, Li KC, Chan HL. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol 2013; 12: 256-262.
- Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol 2013; 19: 6911-6918.
- Vajro P, Mandato C, Licenziati MR, Franzese A, Vitale DF, Lenta S, Caropreso M, Vallone G, Meli R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J Pediatr Gastroenterol Nutr 2011; 52: 740-743.
- Yadav H, Lee JH, Lloyd J, Walter P, Rane SG. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J Biol Chem 2013; 288: 25088-25097.
- Plaza-Diaz J, Gomez-Llorente C, Abadia-Molina F, Saez-Lara MJ, Campaña-Martin L, Muñoz-Quezada S, Romero F, Gil A, Fontana L. Effects of Lactobacillus paracasei CNCM I-4034, Bifidobacterium breve CNCM I-4035 and Lactobacillus rhamnosus CNCM I-4036 on hepatic steatosis in Zucker rats. PLoS One 2014; 9: e98401.
- Du J, Ma YY, Yu CH, Li YM. Effects of pentoxifylline on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol 2014; 20: 569-577.
- Schrieber SJ, Hawke RL, Wen Z, Smith PC, Reddy KR, Wahed AS, Belle SH, Afdhal NH, Navarro VJ, Meyers CM, Doo E, Fried MW. Differences in the disposition of silymarin between patients with nonalcoholic fatty liver disease and chronic hepatitis C. Drug Metab Dispos 2011; 39: 2182-2190.
- Yao J, Zhi M, Minhu C. Effect of silybin on high-fat-induced fatty liver in rats. Braz J Med Biol Res 2011; 44: 652-659.
- Solhi H, Ghahremani R, Kazemifar AM, Hoseini Yazdi Z. Silymarin in treatment of non-alcoholic steatohepatitis: A randomized clinical trial. Caspian J Intern Med 2014; 5: 9-12.
- Polyak SJ, Ferenci P, Pawlotsky JM. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 2013; 57: 1262-1271.
- Kim M, Yang SG, Kim JM, Lee JW, Kim YS, Lee JI. Silymarin suppresses hepatic stellate cell activation in a dietary rat model of non-alcoholic steatohepatitis: analysis of isolated hepatic stellate cells. Int J Mol Med 2012; 30: 473-479.
- Xanthakos SA, Kohli R. Treating pediatric nonalcoholic fatty liver disease with cysteamine: is adiponectin the key? J Pediatr 2012; 161: 579-581.
- Dohil R, Meyer L, Schmeltzer S, Cabrera BL, Lavine JE, Phillips SA. The effect of cysteamine bitartrate on adiponectin multimerization in non-alcoholic fatty liver disease and healthy subjects. J Pediatr 2012; 161: 639-645.
- Dohil R, Schmeltzer S, Cabrera BL, Wang T, Durelle J, Duke KB, Schwimmer JB, Lavine JE. Enteric-coated cysteamine for the treatment of paediatric non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2011; 33: 1036-1044.
- Oberg HH, Juricke M, Kabelitz D, Wesch D. Regulation of T cell activation by TRL ligands. Eur J Cell Biol 2011; 90; 582-592.
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140: 805-820.
- Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010; 33: 861-868.
- Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I. High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes 2008; 57: 3090-3098.
- Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease update. Hepatology 2008; 48: 322-335.
- Isogawa M, Robek MD, Furuichi Y, Chisari FV.Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 2005; 79: 7269-7272.
- Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O, Okamura H, Fujimoto J, Akira S, Nakanishi K. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol 2001; 166: 2651-2657.
- Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010; 129: 363-374.
- Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003; 37: 1043-1055.
- Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, Szabo G. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 2011; 300: G433-G441.
- Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010; 139: 323-334.
- Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasime activation in mice. Hepatology 2013; 57: 577-589.
- de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrígues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr 2002; 21: 219-223.
- Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like recepto-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 2003; 278: 37041-37051.
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science 1999; 284: 1362-1365.
- Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013; 145: 574-582.
- Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, Gunyuzlu PL, Haws TF, Kassam A, Powell F, Hollis GF, Young PR, Mukherjee R, Burn TC. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 2002; 290: 35-43.
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E; NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015; 385: 956-965.
- Singh S, Khera R, Allen AM, Murad MH, Loomba R. Comparative effectiveness of pharmacological interventions for non-alcoholic steatohepatitis: A systematic review and network meta-analysis. Hepatology 2015; 62: 1417-1432.
- Lee J, Hong SW, Rhee EJ, Lee WY. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab J 2012; 36: 262-267.
- Kelly AS, Rudser KD, Nathan BM, Fox CK, Metzig AM, Coombes BJ, Fitch AK, Bomberg EM, Abuzzahab MJ. The effect of glucagon-like peptide-1 receptor agonist therapy on body mass index in adolescents with severe obesity: a randomized, placebo-controlled, clinical trial. JAMA Pediatr 2013; 167: 355-360.
- Jinmi Lee, Seok-Woo Hong, Eun-Jung Rhee, Won-Young Lee. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab J 2012; 36: 262-267.
- Gupta N, Mells J, Dunham R, Grakoui A, Handy J, Saxena N, Anania F. Glucagon-Like Peptide-1 Receptor Is Present on Human Hepatocytes and Has a Direct Role in Decreasing Hepatic Steatosis In Vitro by Modulating Elements of the Insulin Signaling Pathway. Hepatology 2010; 51: 1584-1592.
- Patel KR, White FV, Deutsch GH. Hepatic steatosis is prevalent in stillborns delivered to women with diabetes mellitus. J Pediatr Gastroenterol Nutr 2015; 60(2): 152-158.
- Brumbaugh DE, Tearse P, Cree-Green M, Fenton LZ, Brown M, Scherzinger A, Reynolds R, Alston M, Hoffman C, Pan Z, Friedman JE, Barbour LA. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr 2013; 162(5): 930-936.
- Modi N, Murgasova D, Ruager-Martin R, Thomas EL, Hyde MJ, Gale C, Santhakumaran S, Doré CJ, Alavi A, Bell JD. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res 2011; 70(3): 287-291.
- Ugalde-Nicalo PA, Schwimmer JB. On the origin of pediatric nonalcoholic Fatty liver disease. J Pediatr Gastroenterol Nutr 2015; 60(2): 147-148.
Correspondencia: Fernando Sarmiento Quintero
Correo electrónico: fsarmientoq@unal.edu.co, fesarq@hotmail.com
Acta Gastroenterol Latinoam 2016;46(3): 246-264
