
Carla Pennella,1 Andrea Bosaleh,2 Adriana Rose,1 Pedro Zubizarreta,1 Walter Cacciavillano1
1 Servicio de Hematología y Oncología.
2 Servicio de Patología.
Hospital de Pediatría S.A.M.I.C “Prof. Dr. Juan P. Garrahan”. Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2020;50(2):162-168
Recibido: 02/09/2018 / Aprobado: 28/08/2019 / Publicado en www.actagastro.org el 29/06/2020

Summary
Although pancreatoblastoma (PB) is a rare tumor, it is the most common malignant pancreatic tumor in children. Clinic presentation is insidious, so early diagnostic suspicion allows timely therapy. We reported 3 cases of PB treated at our center. The first two cases achieved complete disease response after full tumor resection. The first one is in complete remission at 7 months after chemotherapy. The second patient is in second complete remission at 206 months after diagnosis and 128 months after metastatic relapse. The third case died from disease progression 61 months after the initial metastatic unresectable tumor. Histology, clinical features and treatment options are discussed along with presentation of the cases.
Key words. Pancreatoblastoma, pancreatic tumor, children, rare tumors, treatment.
Pancreatoblastoma. Reporte de casos y revisión bibliográfica
Resumen
Si bien el pancreatoblastoma es considerado un tumor raro, es el tumor maligno pancreático más frecuente en pediatría. Su clínica es insidiosa, por lo que la sospecha diagnóstica precoz permite una terapéutica oportuna. Reportamos tres casos de pancreatoblastoma tratados en una institución. Los primeros dos casos se encuentran libre de enfermedad luego de la resección completa del tumor. El primero se encuentra en remisión completa luego de siete meses de finalizado el tratamiento. El segundo paciente está en segunda remisión completa, luego de 206 meses desde el diagnóstico y 128 meses luego de la recaída metastásica. El tercer caso falleció por progresión de la enfermedad, luego de 61 meses desde el diagnóstico inicial de enfermedad metastásica irresecable. Se revisan las características histológicas, clínicas y las opciones de tratamiento junto con la presentación de los casos.
Palabras claves. Pancreatoblastoma, tumores pancreáticos, tumores raros, tratamiento.
Abbreviations
VPB: Pancreatoblastoma.
AFP: Alpha-fetoprotein.
CT: Computer tomography.
Introducción
Pancreatoblastoma (PB) is a rare exocrine pancreatic tumor of childhood, which can sporadically affect adults. Although it comprises 0.5% of pancreatic tumors,1 is the most common pancreatic malignant tumor of childhood.2, 3 Essential for accurate histological diagnosis are acinar differentiation and the presence of squamoid nests.4
Clinical onset is mainly insidious or asymptomatic, driving a frequent advanced initial staging (III, IV according to TNM).5 Most of times is associated with elevation of Alpha-fetoprotein (AFP).6, 7 Surgery may be curative when complete resection is possible, but non resectable or metastatic disease has a dismal outcome.2, 8
Less than 200 cases have been reported in the literature.1 The knowledge about this rare disease is important due to the relevance of an early diagnosis. The objective of this paper is to report three cases treated over 30 years in the care center “Hospital de Pediatría S.A.M.I.C. Prof. Dr. Juan P. Garrahan» and a review of the literature.
Case 1
A palpable epigastric mass was detected in a five-year old male patient during health control. The child complained of abdominal pain without any other accompanying symptom. Ultrasound study showed a pancreatic tumor.
Initial CT-Scan disclosed a tumor of lobed contours at the pancreatic body, with heterogeneous density, hypo-dense areas and calcifications that contacted with the liver without clear-cut limits and volume of 145 cm3. Superior mesenteric vein showed tumor invasion. (Figure 1); A-FP serum level was 239 ng/ml (normal range 8.5 ± 5.5 ng/ml). There was no evidence of disease in thoracic and brain CT-scan.
Figure 1. Abdomen and pelvis CT scan before treatment. (A) Axial plane and (B) coronal plane, highlight lack of preservation of flat planes between tumor mass and liver. (C) Coronal plane in arterial time, heterogeneous enhancement is observed with hyper-dense area in segment III of liver in contact with tumor mass.

Percutaneous trucut biopsy was made, and the pathology report informed pancreatoblastoma, stage IIA according to TNM.
Due to the inability to assure a surgical complete resection, neo-adjuvant chemotherapy with cisplatin/doxorubicin was delivered, following SIOPEL 3 guidelines (Liver Tumor Strategy Group of the International Society of Pediatric Oncology). Post third cycle evaluation showed AFP 15ng/ml and CT-scan with very good partial response: tumor volume of 9.7 cm3, superior mesenteric invasion and splenic veins compression (Figure 2). After 4th neo-adjuvant chemotherapy cycle, surgical resection was performed with free microscopic margins. Therapy was completed after two cycles of adjuvant chemotherapy without evidence of residual disease. A transitory elevation of AFP was seen after surgery. Currently free of disease +10 months’ post-diagnosis.
Figure 2. Abdomen and pelvis CT scan after third neo-adjuvant chemotherapy cycle. (A) Axial plane and (B) coronal plane highlight heterogeneous formation with hypo-dense central area and calcifications that partially invades superior mesenteric vein. (C) Coronal plane in arterial time shows resolution of image in segment III of liver.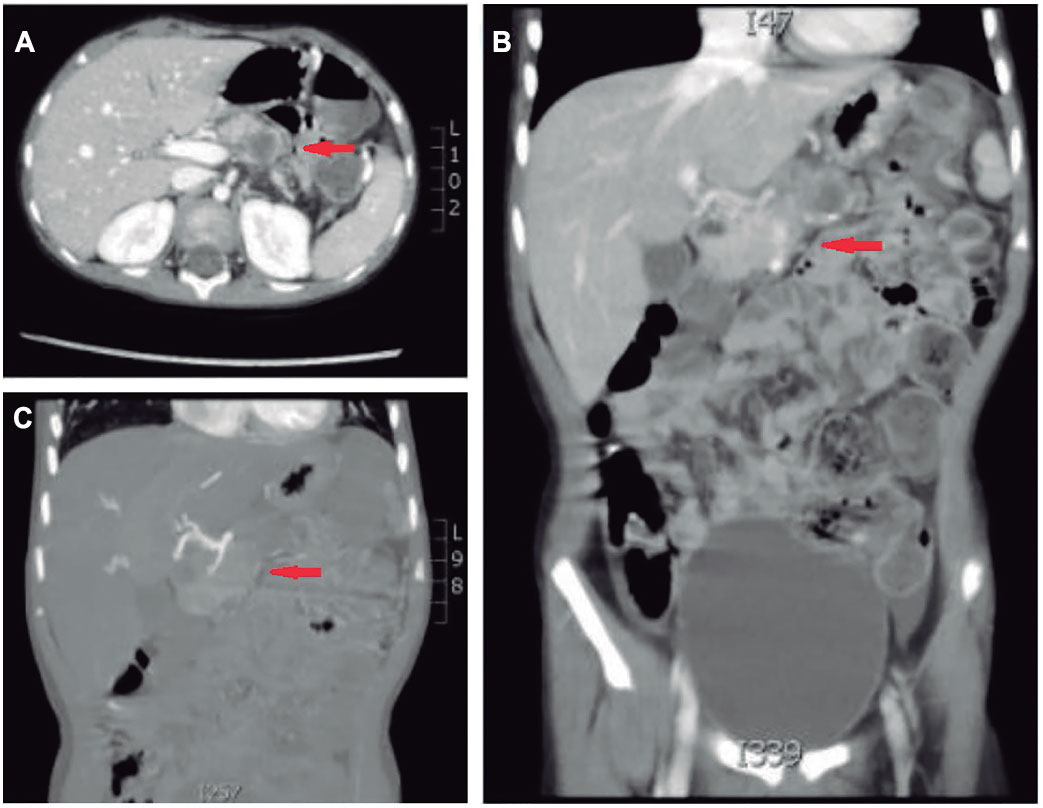
Case 2
A six-year-old female patient complained of pain and abdominal distension during the last 3 months. Abdominal ultrasound showed a pancreatic well-defined heterogeneous mass with multiple necrotic areas of 115 x 74 mm. However, a precise localization of the tumor was not possible based on images. Initial AFP serum levels of 615 ng/ml. Thorax and Brain CT-Scan without noteworthy findings.
A distal pancreatectomy was performed and pathology analysis reported a Pancreatoblastoma, stage IIA (Figure 3).
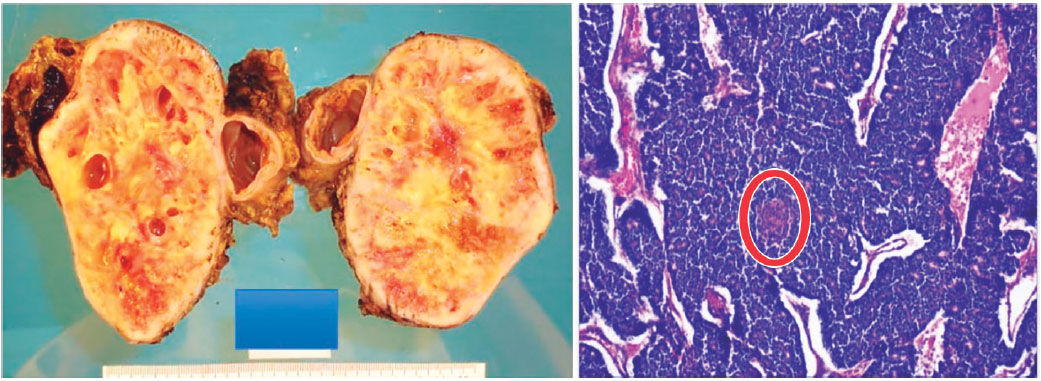
Figure 3. Macroscopic and microscopic pathology (case 2). Cystic-solid tumor, well-defined mass. Histologically solid pattern, tubulo-acinar with squamoid morula (circular mark). Hematoxylin-eosin coloration 100X.
Post-surgery serum level of AFP was 73.8 ng/ml and AFP returned to normal after six months. Six years later AFP level increased to 210 ng/ml and CT-scan showed local and metastatic recurrence at liver segment VIII. New tumor and metastasis excision was performed, with free microscopically margins. Post-surgical AFP level was 7.2 ng/ml. Adjuvant chemotherapy was started delivering 5 cycles of carboplatin/doxorubicin + cisplatin (SIOPEL 2 guidelines). A Second Complete Remission was achieved with normal levels of AFP. Currently the patient is free of disease +206 months (17 years old) post-diagnosis.
Case 3
11-year-old male patient consulted for asthenia and weight loss during the previous month, palpable tumor mass and peripheral lymph nodes. Initial CT-scan showed at liver segment VI a hypo dense solid image of 30mm in the greatest diameter, at caudate lobe image of 40mm in the greatest diameter, and at pancreas head a heterogeneous formation of 44 x 40mm with hypo dense areas with contrast enhancement, associated with retroperitoneal lymphadenopathies. Brain and thoracic CT-scan and bone technetium-scan without findings. AFP serum level was < 3 ng/ml. A biopsy of a left supraclavicular lymphadenopathy reported metastatic pancreatoblastoma.
Systemic chemotherapy was started with cisplatin-doxorubicin (SIOPEL 3 guidelines). Six cycles were delivered with partial response by CT-Scan: residual image at pancreas head of 5.5 cm3 remained. Residual tumor excision was performed, but during surgery hepatic metastases and retroperitoneal lymph nodes where observed and confirmed by biopsy. Second line treatment was started with carboplatin/etoposide for 6 cycles achieving second remission. One month after the end of treatment, a new lesion of 1.8 cm3 was seen in the uncinated process of pancreas. Tumor excision was performed but biopsy showed lymphatic lumen and peri neural neoplastic involvement. There was no other evidence of macroscopic disease and a close follow-up ensued. Three years later the patient suffered a third recurrence with lesions in pancreas tail and bilateral multiple pulmonary metastases. The AFP level was 10 ng/ml. A third line of treatment was started by 3 cycles of cyclophosphamide/topotecan and irinotecan 2 cycles with progressive disease. The patient survived with palliative care 61 months post-diagnosis, and 8 months after last relapse.
Discussion
TPB is considered a rare tumor of childhood. According to the SEER (Surveillance, Epidemiology and End Results) the annual incidence of pancreatic tumors is 0.191 per million, in the population between 0-19 years-old.8 Argentine pediatric cancer Registry (ROHA) has registered 27 patients aged 0-15 years-old with pancreatic tumors from 2000 to 2017: 4 PB.
Is commonly diagnosed in the first decade of life (average age of 2.4-5 years-old)1, 2, 7, 9 and is more frequent in males (1.3-2:1).1, 7 Abdominal pain and palpable mass are the main initial symptoms. They are large tumors (7-18 cm in diameter). Due to soft consistency rarely generates symptoms of duodenal obstruction.10 75% of cases are associated with AFP serum levelsincreasing,7 showing the embryonic origin as hepatoblastoma. Findings on imaging studies are suggestive: presence of a large well-defined mass, multilobulated, heterogeneous with necrosis or calcification and septa which enhance in CT-scan. The ultrasound demonstrates mixed echogenicity with hypo-echoic areas corresponding to necrosis. Vascular invasion makes them more aggressive. At onset one third of patients has distant metastases being liver the most common site,9 then lung, bone, mediastinum, and lymph nodes.
Although it was a small number of patients, our cases were somewhat older than usual, and clinical data and initial images were consistent with the literature.
PB originates in persistent pluripotent embryonic cells and histologically resembles the acinar fetal pancreatic tissue incompletely differentiated at week 8 of gestation.11 Histologically is characterized by an acinar component with squamous cell differentiation but can exhibit in two thirds endocrine and focal ductal differentiation.4, 12 The solid hyper cellular areas consist of nests of polygonal cells alternating with areas of acinar differentiation forming luminal spaces with small glandular luminal. The stroma can be from paucicellular to hypercellular.13 The characteristic «squamoid corpuscles» vary in shape of flat epithelioid cells islets to frank keratinization.12
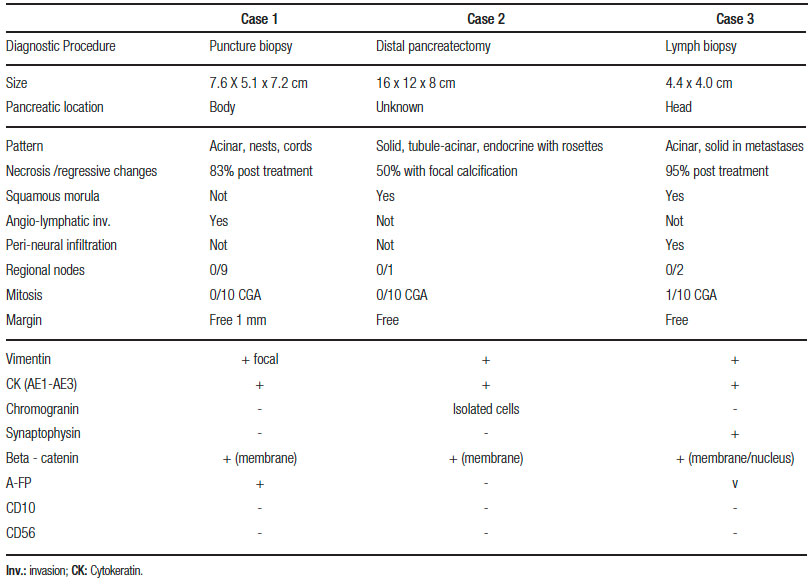
Approximately 90% shows acinar differentiation stained by immunohistochemistry of pancreatic enzymes such as trypsin, chymotrypsin, Alpha-1-antitrypsin or lipase. The epithelial components can be positive for A-FP associated with serum elevations. In two thirds of cases there may be endocrine differentiation with positive staining for chromogranin, synaptophysin, or specific-neuronal enolase.12 The nuclear or cytoplasmic Beta-catenin is especially observed in 80% of the squamous corpuscles and correlates with molecular alterations of the path of APC/beta-catenin in 40-60% of the sporadic PB13 and forms of congenital presentation (associated with Beckwith – Wiedemann syndrome and Familial Adenomatous Polyposis). 86% have alterations at chromosome 11p15.5.4, 13 These alterations are associated with other embryonic tumors such as hepatoblastoma, suggesting a common genetic origin. Pathologic findings of reported patients are shown in Table 1.
Table 1. Pathologic findings of reported patients.
The best treatment option is surgery and a complete resection may be curative. The invasion of portal vein or hepatic artery, metastatic disease and invasion of local vascular structures contraindicate initial surgical intervention. In these cases, neo-adjuvant chemotherapy is used.2, 8 Many useful agents were reported, but because of the similarity with hepatoblastoma PLADO scheme is recommended (cisplatin 80 mg/m2 and doxorubicin 60 mg/m2). The AFP can be used as a parameter of tumor response.2, 8 In patients without response to neo-adjuvant treatment, incomplete resection or local recurrence, radiation therapy can be used as a therapeutic option.8, 14 There is no standard treatment regimen for metastatic pancreatoblastoma.14
The overall survival at 5-year is 63.7% (48–79.4%) in the present largest series1, 7 and the main prognostic factor is achieving complete surgical resection.1, 7 Follow-up guidelines where not established yet. It is suggested periodic physical examinations, images, and AFP monitoring.2 The mean follow-up of our patients was 92 (10-206) months. The first two cases achieved complete disease response given the full macroscopic and microscopic tumor resection.
Since pancreatoblastoma is a rare pediatric pathology, it is of great importance a multidisciplinary approach, with the presence of oncologists, pathologists, surgeons and imaging specialists, to achieve a correct diagnosis and management.
Financial support. Not Received.
References
- Dhebri AR, Connor S, Campbell F, Neoptolemos JP. Diagnosis, treatment and outcome of pancreatoblastoma. Pancreatology 2004; 4: 441-453.
- Glick RD, Pashankar FD, Pappo A, Laquaglia MP. Management of pancreatoblastoma in children and young adults. J Pediatr Hematol Oncol 2012; 34 (2): S47-S50.
- Chung EM, Travis MD, Conran RM. Pancreatic tumors in Children: radiologic-Pathologic Correlation. Radio Graphics 2006; 26: 1211-1238.
- Hackeng WM, Hruban RH, Offerhaus GJ, Lodewijk AAB. Surgical and molecular pathology of pancreatic neoplasms. Diagn Pathol 2016; 11 (1): 47.
- Gospodarowicz MK, Wittekind C, Brierley JD. TNM Classification of Malignant Tumors, 8th Edition (UICC). Oxford: Wiley, 2016.
- Iseki M, Suzuki T, Koizumi Y, Hirose M, Laskin WB, Nakazawa S, Ohaki Y. Alpha-fetoprotein-producing pancreatoblastoma. A case report. Cancer 1986; 57 (9): 1833-1835.
- Bien E, Godzinski J, Dall’igna P, Defachelles AS, Stachowicz-Stencel T, Obrach D, Bisogno G, Cecchetto G, Warmann S, Ellerkamp V, Brennan B, Balcerska A, Rapala M, Brecht I, Schneider D, Ferrari A. Pancreatoblastoma: a report from the European cooperative study group for pediatric rare tumors (EXPeRT). Eur J Cancer 2011; 47 (15): 2347-2352.
- Dall’igna P, Cecchetto G, Bisogno G, Conte M, Lelli Chiesa P, D’Angelo P, De Leonardis F, De Salvo G, Favini F, Ferrari A; on Behalf of the TREP Group. Pancreatic tumors in children and adolescents: the Italian TREP project experience. TREP Group. Pediatr Blood Cancer 2010; 54 (5): 675-680.
- Rojas Y, Warneke CL, Dhamne CA, Tsao K, Nuchtern JG, Lally KP, Vasudevan SA, Hayes-Jordan AA, Cass DL, Herzog CE, Hicks MJ, Kim ES, Austin MT. Primary malignant pancreatic neoplasms in children and adolescents: a 20 year experience. J Pediatr Surg 2012; 47 (12): 2199-2204.
- Low G, Panu A, Millo N, Leen E. Multimodality imaging of neoplastic and non-neoplastic solid lesions of the pancreas. Radiographies 2011; 31 (4): 993-1015.
- Horie A, Yano Y, Kotoo Y, Miwa A. Morphogenesis of pancreatoblastoma, infantile carcinoma of the pancreas: report of two cases. Cancer 1977; 39 (1): 247-254.
- Klimstra DS, Longnecker D. Pancreatoblastoma. En: Hamilton SR., Aaltonen LA, eds. World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of the Digestive System. IARC Press Lyon 2000: 244-245.
- Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee J-H, Yeo CY, Cameron JL, Hruban RH. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 2001; 159 (5): 1619-1627.
- Zheng J, Zhang H, Sun Y, Sun B. CT-guided radiofrequency ablation following high-dose chemotherapy of a liver-metastasizing pancreatoblastoma with thrombus in the portal vein. Pediatr Radiol 2013; 43: 1391-1395.
Correspondencia: Carla Pennella
Combate de los Pozos 1881 (C 1245 AAM). Ciudad Autónoma de Buenos Aires, Argentina
Tel.: (54)911-4177882
Correo electrónico: carlapen@gmail.com
Acta Gastroenterol Latinoam 2020;50(2):163-168
