Francisco Cammarata-Scalisi,1, 2 Dianora Araque,1 Frances Stock,3 Mercedes Molina,4 Raúl Rodríguez,4 Greicy Vázquez,4 Graciela Cammarata-Scalisi,2 María Elena Cammarata-Scalisi2
1 Unidad de Genética Médica, Departamento de Puericultura y Pediatría, Universidad de Los Andes, Mérida, Venezuela.
2 Colaborador de la Asociación Merideña para el Síndrome de Down, Venezuela.
3 Unidad de Oncología Pediátrica, Instituto Autónomo Hospital Universitario de Los Andes, Mérida, Venezuela.
4 Postgrado de Puericultura y Pediatría, Hospital Dr Luis Razetti, Barinas, Venezuela.

Resumen
La trisomía X es una anomalía cromosómica sexual que puede presentarse en forma de mosaico. No es infrecuente y la mayoría de los casos no son diagnosticados. La prevalencia se ha establecido en una de cada mil mujeres. Se caracteriza clínicamente por la estatura alta, microcefalia, hipertelorismo, anomalías congénitas, retraso motor y del lenguaje. La asociación entre la trisomía X y las malformaciones gastrointestinales es extremadamente infrecuente. Se presenta un caso de trisomía X en mosaico con obstrucción gástrica ampliando el espectro clínico de esta entidad y haciendo énfasis en su desconocida patogénesis.
Palabras claves. Trisomía X, mosaico, obstrucción gástrica, patogénesis.
Gastrointestinal obstruction in the mosaic trisomy X
Summary
Trisomic X is a sex chromosomal abnormality that may be presented in mosaic. This is not extremely rare, the majority of cases go undiagnosed. The prevalence has been established to 1/1000 females. It is clinically characterized by tall stature, microcephaly, hypertelorism, congenital abnormalities, and motor and language delays. The association between the trisomic X and gastrointestinal malformations is extremely rare. We report a case of mosaic trisomic X with gastric obstruction expanding the clinical spectrum of this entity and emphasizing its unknown pathogenesis.
Key words. Trisomic X, mosaic, gastric obstruction, pathogenesis.
La trisomía del cromosoma X presenta una prevalencia a 1 de cada 1.000 mujeres y es con frecuencia no diagnosticada. Esta aneuploidía consiste en tres cromosomas X, 47,XXX, y puede exhibirse en forma de mosaico cuando se evidencian dos grupos celulares, uno con tres cromosomas X y otro con dos normal 47,XXX/46,XX en un mismo individuo. Fue descripta por primera vez por Jacobs en 1959 en una mujer con infertilidad.1 Entre los diversos hallazgos fenotípicos se encuentran estatura alta, hipertelorismo, pliegues epicánticos, clinodactilia,2, 3 cardiopatía congénita y alteraciones genitourinarias, entre otras.2 Los estudios de neuroimagen evidencian disminución significativamente menor del volumen cerebral, lo que puede asociarse con dificultad en el aprendizaje,2 retardo en el desarrollo motor y del lenguaje.2, 3 Pueden cursar timidez, alteraciones en las relaciones interpersonales, así como en el manejo del estrés,2 ansiedad, trastornos del humor, hiperactividad con déficits de atención,3 y autismo.4 En algunos casos experimentan retardo en la menarquía2, 5 y falla ovárica precoz.2, 3 La mortalidad es significativamente mayor en las mujeres con la entidad de la misma edad.2
Esta aneuploidía se produce por la no disyunción en la meiosis I y es en el 90% de los casos aproximadamente de origen materno y en el 10% paterno. Su detección prenatal presenta importantes dificultades al momento de emitir asesoramiento genético. El diagnóstico postnatal puede verse dificultado porque la mayoría de los casos tienen un fenotipo normal y no manifiestan importantes alteraciones clínicas.6 El objetivo de este informe es presentar un caso clínico de trisomía X en mosaico con diagnóstico prenatal de obstrucción gástrica, ampliando el espectro clínico de esta entidad genética, haciendo énfasis en lo desconocido de su etiopatogenia.
Caso clínico
Preescolar femenina de dos años y un mes de edad, evaluada por presentar diagnóstico prenatal de obstrucción gástrica por estudio de ecografía fetal y trisomía del cromosoma X por amniocentesis.
Los padres son sanos, no consanguíneos. El padre de 28 años y la madre de 22 años de edad para el momento de la concepción. No se presentan casos similares en ambas familias de los progenitores. La paciente es producto de primigesta, embarazo controlado a partir del mes de gestación, complicado con preeclampsia e hipomotilidad fetal al final de la misma. Se realizó diagnóstico prenatal por estudio de ecografía morfogenética de doble burbuja a nivel abdominal. Se realizó estudio de amniocentesis que evidenció 47,XXX, en 20 metafases estudiadas.
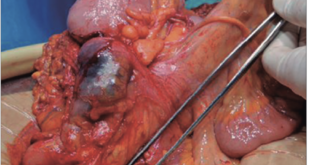
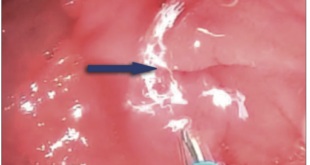
Fue obtenida por cesárea segmentaria a las 35 semanas de gestación, la presentación fue cefálica, el peso al nacer: 1.400 g (P <3) SDS -4,6; talla al nacer: 44 cm (P <3); SDS -4,1. Respiró y lloró al nacer, el test de Apgar de 7 y 9 puntos al primer y quinto minuto respectivamente, no presentó cianosis ni ictericia. Fue intervenida quirúrgicamente por obstrucción gástrica (membrana prepilórica) durante el período neonatal permaneciendo hospitalizada durante 27 días con evolución clínica favorable.
Presentó hipotonía generalizada, retardo del desarrollo psicomotor con inicio de la marcha a los 25 meses de edad y del lenguaje emitiendo las primeras palabras a los 23 meses, por lo que se indicaron terapias de estimulación motora y lenguaje. Recibió tratamiento por anemia severa a los 8 meses de edad con respuesta satisfactoria.
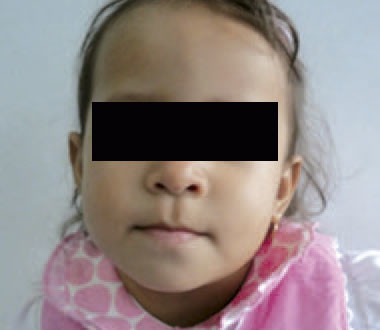
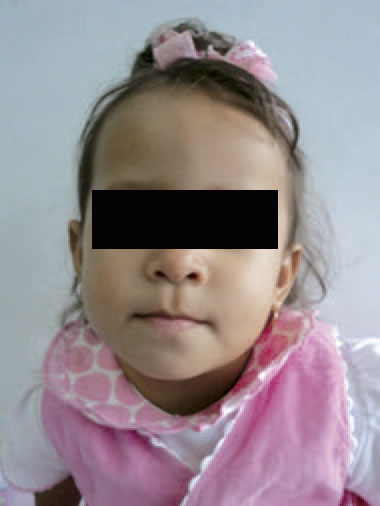
Examen físico: realizado a los dos años y un mes. Talla: 79 cm (P >3 – ≤ 10); SDS -1,9; peso: 9,7 kg (P >3 – ≤ 10); SDS -1,6; CC: 46,1 cm (P 10-25). Normocefalia, el cabello es escaso, la región frontal es prominente, con cejas escasas, las hendiduras palpebrales son horinzontalizadas, narinas antevertidas y la punta nasal es bulbosa, el filtrum bien dibujado y el labio superior es fino (Figura 1). El paladar es ojival, con malposición y malaoclusión dentaria. Los pabellones auriculares con leve rotación posterior y en asa. El cuello es corto, el tórax es simétrico con murmullo vesicular audible sin agregados y los ruidos cardiacos sin soplos. El abdomen sin evidencia de organomegalias, los genitales externos son normo-configurados, con estadio de maduración Tanner I. En las extremidades se presenta un pliegue transverso único en mano izquierda y el 5º dedo de pie izquierdo incurvado. En piel se evidencia cicatriz en flanco derecho y región supraumbilical. A nivel neurológico, la paciente se muestra vígil, introvertida y colaboradora. Los reflejos osteotendinosos están presentes y la fuerza muscular conservada.
El estudio citogenético postnatal fue 46,XX/47,XXX, 87,5% de células con la trisomía del X, con 40 metafases estudiadas por técnica convencional de banda G. La evaluación cardiovascular y la ecografía abdomino-renal fue normal.
Figura 1. Rostro de la paciente que no evidencia dismorfia facial de importancia.
Discusión
La presencia de obstrucciones gastrointestinales en la trisomía del cromosoma X es extremadamente infrecuente. Rolle y col presentan una paciente con diagnóstico prenatal de atresia duodenal por ecografía, evidenciándose estómago y parte superior del duodeno dilatado (fenómeno de doble burbuja) y la alteración citogenética por estudio de amniocentesis.7 La alteración fue confirmada y tratada postnatalmente con duodeno-duodenostomía. Por su parte, Trautner y col8 describen el primer caso de atresia del yeyuno en una paciente con trisomía del X e igualmente diagnosticado prenatalmente y confirmado al nacer, con polihidramnios en ambos casos.7, 8 Posteriormente, se documentó otro con atresia del duodeno asociado con cardiopatía congénita.9 Por lo tanto, el diagnóstico prenatal de trisomía del X, se debe acompañar de un exhaustivo estudio ecográfico que incluya la exanimación de múltiples órganos que puede garantizar un óptimo manejo clínico perinatal de identificarse alguna malformación congénita.7, 9 El caso presentado constituye el cuarto con obstrucción gastrointestinal según lo revisado en la literatura, el primero a nivel gástrico y de tipo mosaico.
La etiología de la atresia gastrointestinal sigue siendo incierta y entre los posibles eventos se incluyen: los accidentes vasculares en útero, el fracaso en la recanalización de la luz intestinal10, 11 y la compresión mecánica.10, 12 Los estudios realizados por Reader y col en modelo animal de ratón con el receptor del factor de crecimiento de fibroblastos 2IIIb (Fgfr2IIIb-/-) y atresia intestinal, no se asoció a defectos en el desarrollo de la notocorda o alteraciones en la expresión del gen Sonic Hedgehog, Shh.10 Esta hipótesis surgió de las observaciones clínicas de las anomalías de la notocorda con atresia intestinal, así como el desarrollo alterado de la notocorda observado en 80% del modelo animal farmacológico de atresia intestinal producido por la exposición intrauterina a la adriamicina, un agente antineoplásico conocido por interferir con el crecimiento celular. Igualmente, se ha observado en los embriones una expresión ectópica del gen Shh, que se postuló como causa en la formación de la atresia intestinal. El modelo genético mutante homocigótico Fgfr2IIIb-/- desarrolló atresia de colón con 100% de penetrancia y 42% en duodeno. Los defectos en Fgfr2IIIb nulos incluyen una serie de alteraciones esqueléticas, en la piel y anomalías viscerales derivadas de la incapacidad del factor de crecimiento fibroblástico 10, FGF10, expresado en el mesénquima y activa al Fgfr2IIIb, en el epitelio en el desarrollo del intestino. La formación de las atresias en colon y duodeno en embriones Fgfr2IIIb-/- incluyen un aumento en la muerte celular y reducción en la proliferación endodérmica, eventos que preceden a su formación. Ambos procesos se observan primero en el día embrionario 10,5 y la involución del segmento del intestino atrésico se completa el día 13,5.10, 13
Se han descripto formas de atresia intestinal familiar y asociaciones sindromáticas, sin embargo, como ya se hizo referencia se desconoce algún vínculo genético específico establecido. El FGF10 es un regulador del crecimiento normal y de formación del duodeno y se encuentra activo en la etapa tardía del desarrollo del mismo, los modelos fgf10-/- demostraron atresia duodenal presentando con fallo en la continuidad luminal y dilatación proximal. Este fenotipo se produce con un patrón de herencia autosómico recesivo, con penetrancia incompleta de 38%.11 No obstante, estas factores etiopatogénicos no se encuentran asociados directamente con la trisomía del X.
Gazou y col presentaron un caso de un paciente masculino con retardo mental moderado, sordera neurosensorial, dismorfia facial, estenosis pilórica y obstrucción intestinal,14 el cual se le encontró una deleción de novo de 1,56 Mb en Xq22,3 – q23, la cual abarcó el gen ACSL4 causante del retardo mental no sindrómico y ocho genes vecinos (GUCY2F, NXT2, KCNE1L, TMEM164, MIR3978, AMMECR1, SNORD96B y RGAG1), por lo que la ausencia de algunos de los genes situados en ese segmento pueden afectar el normal desarrollo del tracto gastrointestinal. Aunque esta alteración no corresponde a la trisomía X, se hace referencia a atresias en distintos niveles del tracto digestivo y alteraciones genéticas a ese nivel.
Inicialmente esta descripción busca ampliar el espectro clínico de la trisomía X, a pesar de lo infrecuente, destacando su importancia médica al momento del diagnóstico y el tratamiento. Actualmente se desconocen el o los genes situados en el cromosoma X que pueden producir atresias en distintas áreas del tracto gastrointestinal y cómo éstos afectan el desarrollo embriológico. Estudios moleculares a estos pacientes y modelos genéticos serán necesarios para tratar de elucidar esta hipótesis.
Referencias
- Otter M, Schrander-Stumpel CT, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet 2010; 18: 265-271.
- Li M, Zou C, Zhao Z. Triple X syndrome with short stature: case report and literature review. Iran J Pediatr 2012; 22: 269-273.
- Tartaglia NR, Howell S, Sutherland A, et al. A review of trisomy X (47,XXX). Orphanet J Rare Dis 2010; 5: 8.
- Ali SI, Byrne N, Mulligan A. Autism in association with triple X syndrome. Eur Child Adolesc Psychiatry 2012; 21: 233-235.
- Afshan A. Triple X syndrome. J Pak Med Assoc 2012; 62: 392-394.
- Ben Hamouda H, Mkacher N, Elghezal H, et al. Prenatal diagnosis and prognosis of triple X syndrome: 47,XXX. J Gynecol Obstet Biol Reprod (Paris) 2009; 38: 599-603.
- Rolle U, Linse B, Glasow S, et al. Duodenal atresia in an infant with triple-X syndrome: a new associated malformation in 47,XXX. Birth Defects Res A Clin Mol Teratol 2007; 79: 612-613.
- Trautner MC, Aladangady N, Maalouf E, Misra D. Jejunal atresia in an infant with triple-X syndrome. J Matern Fetal Neonatal Med 2004; 16: 198-200.
- Bağci S, Müller A, Franz A, et al. Intestinal atresia, encephalocele, and cardiac malformations in infants with 47,XXX: Expansion of the phenotypic spectrum and a review of the literature. Fetal Diagn Ther 2010; 27: 113-117.
- Reeder AL, Botham RA, Franco M, et al. Formation of intestinal atresias in the Fgfr2IIIb-/- mice is not associated with defects in notochord development or alterations in Shh expression. J Surg Res 2012; 177: 139-145.
- Kanard RC, Fairbanks TJ, De Langhe SP, et al. Fibroblast growth factor-10 serves a regulatory role in duodenal development. J Pediatr Surg 2005; 40: 313-316.
- Fourcade LM, Mousseau Y, Sauvat F, et al. A New rat model of prenatal bowel obstruction: development and early assessment. J Pediatr Surg 2010; 45: 499-506.
- Nichol PF, Tyrrell JD, Saijoh Y. Retinaldehyde dehydogenase 2 is down-regulated during duodenal atresia formation Fgfr2IIIb-/- mice. J Surg Res 2012; 175: 82-87.
- Gazou A, Riess A, Grasshoff U, et al. Xq22.3-q23 deletion including ACSL4 in a patient with intellectual disability. Am J Med Genet A 2013; 161A: 860-864.
Correspondencia: Francisco Cammarata-Scalisi
Universidad de Los Andes
Mérida 5101, Venezuela.
Tel: 0058 0274 2403208 / Cel: 0058 0424 7296843
Correo electrónico: francocammarata19@gmail.com
Acta Gastroenterol Latinoam 2015;45:129-132