Florencia Yamasato ID· Jorge Daruich ID
Sección Hepatología. Servicio de Gastroenterología. Hospital de Clínicas José de San Martín, Universidad de Buenos Aires.
Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2023;53(2):113-125
Recibido: 17/02/2023 / Aceptado: 28/05/2023 / Publicado online el 30/06/2023 / https://doi.org/10.52787/agl.v53i2.303

Resumen
La hemocromatosis hereditaria o genética engloba a un grupo de trastornos en los que distintas mutaciones generan una sobrecarga de hierro que afecta a diferentes órganos y sistemas. Esta enfermedad librada a su historia natural puede provocar cirrosis, hepatocarcinoma, diabetes mellitus y artropatías entre otras patologías. El compromiso de uno o más componentes del eje hepcidina-ferroportina del sistema del metabolismo del hierro es el común denominador de los distintos tipos de hemocromatosis hereditaria. La mutación del gen HFE C282Y es la más frecuentemente detectada en los pacientes del norte de Europa y su descendencia. El diagnóstico se sustenta en el fenotipo bioquímico, la resonancia magnética que muestra sobrecarga de hierro hepático y la detección de la mutación HFE C282Y, en ausencia de otras comorbilidades. Sin embargo, en Sudamérica la mutación del gen HFE C282Y es poco frecuente, modificando el algoritmo para realizar el diagnóstico de la enfermedad. Los tratamientos de elección que pueden revertir el compromiso de los órganos afectados por la sobrecarga de hierro son las flebotomías o las eritroaféresis.
Palabras claves. Hemocromatosis hereditaria, gen HFE, hepcidina, ferritina, cirrosis, flebotomías.
Hereditary Hemochromatosis
Summary
Hereditary or genetic hemochromatosis is a group of disorders in which different mutations lead to iron overload affecting different organs and systems. This disease left to its natural history can cause cirrhosis, hepatocarcinoma, diabetes mellitus and arthropathies, among other pathologies. The involvement of one or more components of the hepcidin-ferroportin axis of the iron metabolism system is the common denominator of the different types of hereditary hemochromatosis. The HFE C282Y gene mutation is the most frequently detected mutation in Northern European patients and offspring. Diagnosis is based on the biochemical phenotype, magnetic resonance imaging showing hepatic iron overload and detection of the HFE C282Y mutation, in the absence of other comorbidities. However, in South America, the HFE C282Y gene mutation is rare, which changes the algorithm for diagnosing the disease. The treatments of choice, which can reverse the involvement of organs affected by iron overload, are phlebotomies or erythropheresis.
Keywords. Hereditary hemochromatosis, HFE gen, hepcidin, ferritin, cirrhosis, fhlebotomies.
Abreviaturas
HCC: Hepatocarcinoma.
DM: Diabetes mellitus.
HH: Hemocromatosis hereditaria.
DMT1: Sigla en inglés de transportador de metales divalentes 1.
HCP1: Sigla en inglés de proteína transportadora hemo 1.
Fe2+: Hierro en estado ferroso.
Fe3+: Hierro en estado férrico.
FPN1: Ferroportina.
Tf: Transferrina.
NTBI: Sigla en inglés de hierro no unido a la transferrina.
Tfr1: Sigla en inglés de receptores de transferrina 1.
Tfr2: Sigla en inglés de receptores de transferrina 2.
Ft: Ferritina.
HEP: Hepcidina.
HAMP: Sigla en inglés de péptido antimicrobial de hepcidina.
HJV: Hemojuvelina.
SLC40A1: Miembro 1 de la familia de transportadores de solutos 40.
NGS: Sigla en inglés de secuenciación de nueva generación.
FS: Ferritina sérica.
STf: Saturación de transferrina.
AST: Aspartatato aminotransferasa.
ALT: Alanina aminotransferasa.
HHg: Hipogonadismo hipogonadotrófico.
CoQ10: Coenzima Q10.
RM T2*: Resonancia magnética T2 estrella.
Hemocromatosis
La hemocromatosis incluye un grupo de trastornos en los que se produce una acumulación progresiva de hierro en diferentes parénquimas, sobre todo en el hígado. Es una enfermedad sistémica que puede afectar a casi todos los órganos de la economía. Librada a su historia natural, puede provocar cirrosis, hepatocarcinoma (HCC), diabetes mellitus (DM) y artropatías, entre otras patologías.1 Se reconocen tres grandes grupos: a) hemocromatosis hereditaria o genética (HH), b) sobrecarga de hierro adquirida por trastornos diseritropoyéticos o enfermedades hepáticas crónicas y c) administración parenteral de hierro.1
En los últimos años se produjo un gran avance en el conocimiento tanto del metabolismo del hierro como de la fisiopatología de la HH y sus implicancias en el estado oxidativo. En este artículo revisaremos la HH más frecuente, HH tipo I, y los fenotipos observados en Sudamérica.
Metabolismo del hierro
El hierro es un oligoelemento esencial que interviene en múltiples procesos metabólicos. Entre ellos se encuentran la síntesis de hemoglobina, la producción del grupo hemo y de centros ferro-sulfurados (Fe-S). Estos últimos participan como cofactores redox y son esenciales para la función de muchas proteínas y enzimas que intervienen en procesos biológicos fundamentales como la respiración celular, la replicación y reparación de ácidos nucleicos, reacciones metabólicas y mecanismos inmunológicos, entre otras funciones. Por otro lado, a pesar de ser esencial para la vida, el exceso de hierro es tóxico debido a la habilidad de este metal para aceptar y ceder electrones fácilmente; esto origina un aumento de las concentraciones de especies reactivas de oxígeno que dañan a diferentes componentes celulares.2 Debido a que el organismo no tiene la capacidad de eliminar el exceso de hierro, su metabolismo está fuertemente regulado por una serie de mecanismos intracelulares y humorales que controlan la expresión y el catabolismo de diferentes moléculas relacionadas con su absorción, transporte, reserva celular, salida y captación por los tejidos.3
La absorción de hierro es el resultado de un mecanismo complejo que ocurre en el duodeno y el yeyuno proximal. Inicialmente, en el estómago, el hierro hémico es disociado de las hemoproteínas (hemoglobina y mioglobina) y el no hémico es reducido a ferroso (Fe2+).3 En los enterocitos duodenales, el transportador de metales divalentes 1 (DMT1) es el responsable más importante de la absorción del Fe2+. A su vez, el hierro hémico es absorbido en el duodeno por la proteína transportadora hemo 1 (HCP1). Una vez que el hierro ingresa a los enterocitos, su destino dependerá de las concentraciones intracelulares.2
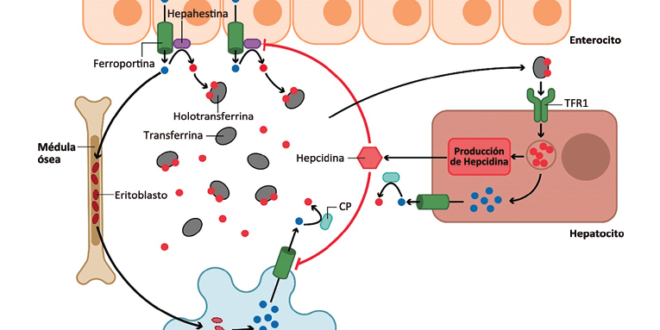
En condiciones en las que existe requerimiento de hierro, este es exportado a la circulación por la proteína transmembrana ferroportina (FPN1), que se encuentra en la membrana basolateral de los enterocitos. La FPN1, única proteína capaz de exportar hierro desde el interior de las células, se encuentra en altas concentraciones en los macrófagos, hepatocitos y enterocitos duodenales. Este micronutriente es transportado por la FPN1 para unirse a la transferrina (Tf). La Tf es una glicoproteína sintetizada principalmente por los hepatocitos, y es la principal transportadora de hierro (Fe3+) en la sangre. En condiciones normales la Tf se encuentra saturada entre el 20 y el 40%. Cuando supera su capacidad se pueden observar formas de hierro no unido a la Tf (NTBI) que inducen una rápida captación del hierro por el hígado y eventualmente por otros órganos, ocasionando toxicidad celular. El hierro unido a la Tf llega a los precursores eritroides, hepatocitos y otras células a través de los receptores de transferrina 1 (Tfr1) y 2 (Tfr2). El complejo hierro-Tf-Tfr1/2 ingresa a la célula donde se libera el metal. A continuación la Tf y el Tfr se disocian para reiniciar un nuevo ciclo de transporte.2 Si no es utilizado, el hierro que ingresa a la célula se almacena unido a la ferritina (Ft), proteína que se encuentra en altas concentraciones en las células del sistema reticuloendotelial y en los hepatocitos, debido a que en su forma libre es altamente tóxico. Cuando es requerido por el organismo, es liberado de la Ft para ser exportado nuevamente a la circulación a través de la FPN1. (Figura 1)
Figura 1. Metabolismo del hierro
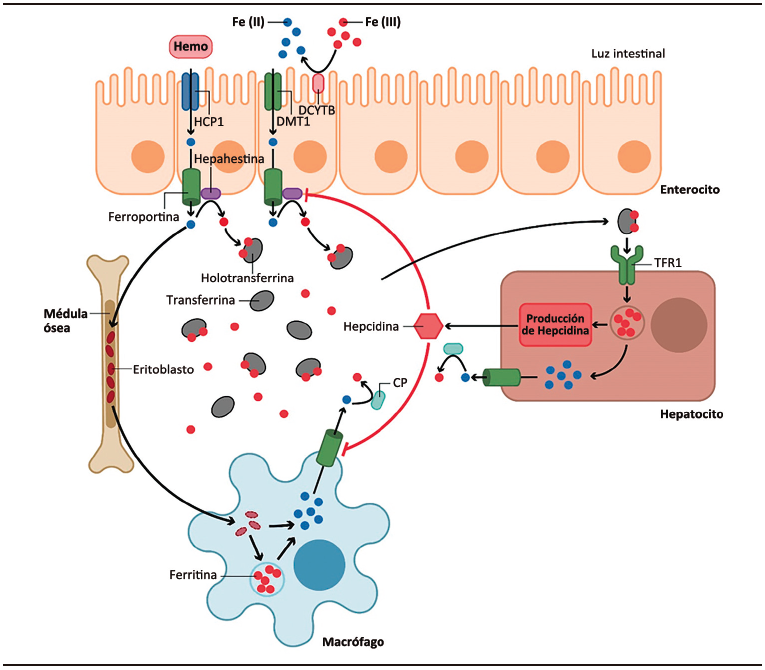
El hierro se absorbe en los enterocitos. La forma no hémica de hierro se absorbe a través del transportador de cationes divalentes 1 (DMT1), luego de que su forma férrica (Fe3+) se transforma en ferrosa (Fe2+) a través de las reductasas duodenales (DCYTB) y otras enzimas. El hierro hémico puede ingresar a los enterocitos a través de transportadores específicos (HCP1). Otra parte del hierro del organismo proviene de la degradación de los glóbulos rojos en los macrófagos. En ambos casos el hierro llega a la sangre a través de la ferroportina (FPN), donde se une a la transferrina (Tf) para poder ser transportado a los diferentes órganos para su utilización, médula ósea y hepatocitos, donde además de ser utilizado para diferentes funciones es almacenado unido a la ferritina. El hígado es el principal regulador de las concentraciones de hierro corporal, ya que en aquellas condiciones donde se saturan los transportadores de transferrina 1 (Tfr1) se estimula la síntesis de hepcidina (HEP), cuya función, entre otras, es estimular la degradación de la FPN para que aumente el hierro en plasma. Diseño de la imagen Lic. Alejandra Yamasato.
Regulación de la homeostasis del hierro
La hepcidina (HEP), péptido antimicrobiano de 25 aminoácidos, es sintetizada fundamentalmente en el hígado. Es la principal reguladora de la homeostasis del hierro. Controla la exportación del hierro hacia el plasma a través de la degradación lisosomal de la FPN1 en los enterocitos, macrófagos y hepatocitos. La HEP disminuye la absorción de hierro cuando esta se encuentra aumentada, y viceversa, cuando la misma está disminuida. Los niveles de HEP son regulados en los hepatocitos por el nivel de hierro circulante y tisular a través de la vía BMP-SMAD que estimula la expresión del gen HAMP (péptido antimicrobial de HEP), que transcribe para la síntesis de HEP. La expresión de la HEP es inhibida por la deficiencia de hierro, la eritropoyesis, la hipoxia, la anemia, y probablemente por otros elementos como la testosterona. El mecanismo más importante por el cual se inhibe la síntesis de HEP es a través de una serin proteasa matriptasa 2, que cliva el complejo BMP-hemojuvelina (HJV), inhibiendo la vía BMP-SMAD.2-4
En relación al metabolismo del hierro, es importante destacar que además de la HEP y la FPN1 intervienen un importante número de genes y péptidos que también pueden ocasionar una sobrecarga o déficit de este micronutriente vital.
Trastornos del metabolismo del hierro: Hemocromatosis hereditaria
La HH engloba a un grupo de trastornos en los que existe alteración del eje HEP-FPN1, en los que la producción de HEP es insuficiente o bien existe una síntesis normal o aumentada, pero con desarrollo de resistencia a la HEP.5-6
Las HH que causan déficit de HEP son de herencia autosómica recesiva.6-7 El tipo más comúnmente observado entre los anglosajones es el asociado a la mutación del gen HFE (HH tipo I), que se encuentra ubicado en el brazo corto del cromosoma 6, siendo la homocigosis C282Y la mutación más frecuentemente descripta (Figura 1). La proteína HFE es una molécula HLA clase I que se encuentra en las membranas celulares en asociación con la β2-microglobulina, que es un cofactor de los Tfr1 y Tfr2. La mutación HFE altera el funcionamiento de los Tfr1 y Tfr2 en respuesta a las concentraciones de hierro. (Figura 2)
Figura 2. Proteína HFE
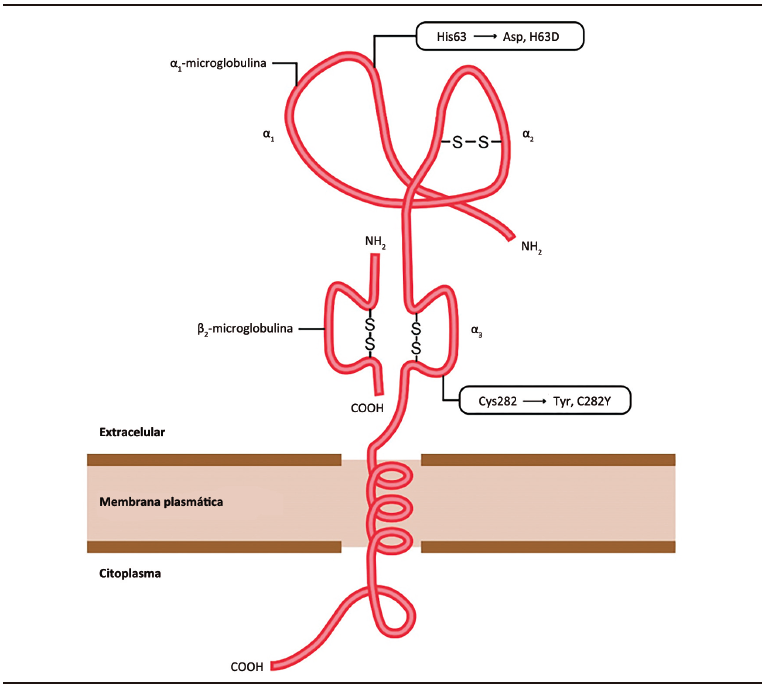
La proteína de transmembrana HFE tiene un papel fundamental en la homeostasis del hierro, facilitando la interacción entre la Tf y su receptor, con la formación de un complejo en el que interviene la β2-microglobulina. Las mutaciones del gen HFE impiden la ubicación correcta de la proteína HFE en la membrana o impiden su asociación con β2-microglobulina y la Tf. Diseño de la imagen Lic. Alejandra Yamasato.
El déficit de HEP también puede ser producto de la mutación de otros genes que transcriben para esta hormona, como HAMP (HH tipo IIb), o genes que aumentan las síntesis de HEP, como HJV (HH tipo IIa) o Tfr2 (HH tipo III).6, 7 (Tabla 1)
Por otra parte, la mutación del gen de la FPN1, llamado SLC40A1, es el único tipo de HH de herencia autosómica dominante. La mutación heterocigota de dicho gen puede producir una pérdida de los transportadores o una resistencia a la HEP (HH tipo IV).
En los últimos años se han descripto modificadores genéticos (polimorfismos de genes) que traducen para proteínas que participan en el metabolismo del hierro y que podrían modificar la penetrancia de los diferentes tipos de HH.3, 8 (Tabla 1)
Tabla 1. Hemocromatosis. Clasificación

Recientemente, la BIOIRON Society recomendó una nueva clasificación de la HH basada en la existencia de otros genes que se encuentran asociados al metabolismo del hierro.9 Esta clasificación divide la sobrecarga de hierro primaria en: 1) asociada al gen HFE; 2) no asociada al gen HFE, relacionada a la mutación de los genes HAMP, TFR2, HJV, SLC40A1 (GOF); 3) formas digénicas asociadas a mutaciones homocigotas/heterocigotas de dos genes diferentes; 4) molecularmente no definida, dado que no se detectan otras secuencias luego de realizar estudios genéticos de rutina. De todas maneras, esta nueva clasificación aún no ha sido aceptada globalmente.
Epidemiología
Los datos epidemiológicos de la HH están sesgados porque los estudios incluyen mayoritariamente pacientes con las mutaciones del gen HFE, debido a las dificultades para analizar otras mutaciones en la práctica clínica. Además, muchos reportes son realizados en centros de referencia y excluyen individuos asintomáticos, lo que podría subestimar la prevalencia de la enfermedad. Con el advenimiento de la secuenciación de nueva generación (NGS) es probable que en los próximos años los datos epidemiológicos en pacientes con HH se modifiquen. Hasta el momento, se estima que la enfermedad afecta a 1 cada 150-220 personas en los descendientes de individuos provenientes del norte europeo.10
En los estudios de prevalencia de las mutaciones realizados en pacientes con HH, el gen HFE está presente en aproximadamente el 80% de los casos en Europa del norte, porcentaje que disminuye en los países del sur europeo. La homocigosis HFE C282Y es la de mayor impacto.6, 10
La prevalencia de la mutación del gen HFE en los pacientes con HH en América del Norte también es elevada, estimándose alrededor del 80%. En Australia, en población blanca no hispana, es cercana al 100%. La heterocigosis C282Y/H63D se ha reportado con una prevalencia de 5,8% y 4,3% en Europa y América del Norte, respectivamente.10, 16
La prevalencia de los genes no HFE en los pacientes con HH tiene una evidencia escasa.
A diferencia de lo descripto en los países anglosajones, en Argentina, un estudio realizado en Buenos Aires en el Hospital de Clínicas José de San Martín en 276 pacientes con HH mostró una prevalencia de 14,29% para la mutación HFE homocigota C282Y y de 9,89% para la heterocigosis C282Y/H63D. A su vez, en la provincia de Misiones, Barreyro F. y col. observaron una prevalencia de 3% de la mutación homocigota C282Y en 34 pacientes con HH.11-12 Otro estudio realizado en Brasil en 51 individuos con HH, también encontró una baja prevalencia de la mutación homocigota C282Y (21,6%) y de la heterocigosis C282Y/H63D (11,7%).13
Manifestaciones clínicas
Los portadores de HH pueden ser asintomáticos o sintomáticos. (Figura 3). Las manifestaciones clínicas aumentan con la edad, en forma paralela al incremento de los depósitos de hierro y a los órganos afectados.
Figura 3. Manifestaciones clínicas
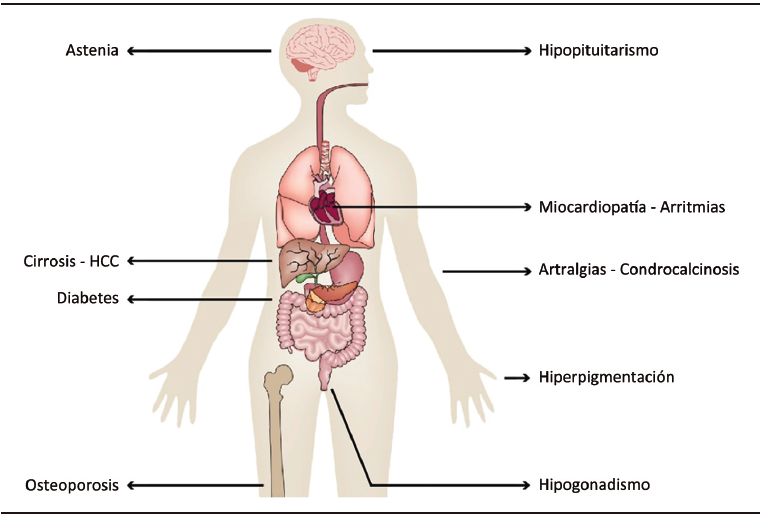
La triada clásica DM, piel bronceada y cirrosis descripta en el siglo XIX raramente se observa en la actualidad debido al diagnóstico en estadios más precoces de la enfermedad. Habitualmente los síntomas se manifiestan en adultos de mediana edad, sobre todo del sexo masculino, con astenia acompañada o no de una leve hipertransaminasemia.14
Publicaciones recientes muestran que en las últimas dos décadas los pacientes portadores de HH tienen una menor sobrecarga de hierro, menor prevalencia de cirrosis y menos manifestaciones extra hepáticas en comparación con los casos detectados previamente, probablemente vinculable al diagnóstico y tratamiento más temprano de la enfermedad.14
En la historia natural de la HH HFE sin tratamiento podemos distinguir una fase preclínica y otra clínica.1,14-15
1- Estadio 0 – Fase preclínica (asintomática): predisposición genética a la sobrecarga, sin alteraciones detectables de la ferritina sérica (FS) o de la saturación de transferrina (STf).
2- Estadio I – Fase preclínica (asintomática): progresión de la sobrecarga de hierro. Se puede detectar elevación de la STf > 45% con FS normal (≤ 300 ng/ml en el varón y ≤ 200 ng/ml en la mujer). En esta fase los individuos continúan asintomáticos, pudiéndose observar sobrecarga de hierro en la resonancia magnética T2 estrella (RM T2*).
3- Estadio II – Fase preclínica (asintomática): sobrecarga de hierro corporal hasta 5 gramos. Se detecta elevación de la FS (> 300 ng/ml en varón y > 200 ng/ml en la mujer) y de la STf (> 45%).
4- Estadio III – Fase clínica: hierro corporal mayor de 5 gramos, con aumento conjunto de la FS (> 300 ng/ml en varón y > 200 ng/ml en la mujer) y de la STf (> 45%). Clínicamente es oligosintomática o sintomática. La calidad de vida puede estar afectada.
5- Estadio IV – Fase clínica: se aprecia daño en diferentes órganos que afecta la calidad de vida y compromete la supervivencia.
Si bien el gen HFE C282Y tiene una elevada prevalencia en la población caucásica, la penetrancia de la enfermedad es baja. Los estudios de seguimiento a largo plazo en los individuos no tratados, portadores del gen HFE C282Y homocigotas/heterocigotas o con mutaciones del gen H63D tienen una penetrancia bioquímica que se ha descripto entre un 57% y un 94% en los varones y en porcentajes menores en las mujeres. En un estudio australiano que incluyó 203 adultos entre 40 y 69 años con HH HFE, en un periodo de seguimiento de 12 años, se observó una penetrancia de enfermedades relacionadas con la sobrecarga de hierro de 28% en varones y 1,2% en mujeres.16
La penetrancia de la enfermedad está modificada por factores genéticos, ambientales y por la edad. En portadores de mutaciones del gen HFE, la concurrencia de mutaciones o polimorfismos en los otros genes que intervienen en el metabolismo del hierro definen una mayor o menor penetrancia. Entre los factores ambientales se destacan el alcoholismo, tabaquismo, enfermedad hepática grasa, sobrepeso y hepatitis crónicas virales, entre otros.1, 17-18
Astenia
Los portadores de HH son habitualmente asintomáticos durante mucho tiempo. Inicialmente, las manifestaciones son inespecíficas y, probablemente, la más frecuente es la astenia. Se suele presentar en los estadios precoces de la enfermedad, al comienzo es leve y con el tiempo puede llegar a ser invalidante. Se desconoce el mecanismo que produce este síntoma.17-18
Injuria hepática
En la gran mayoría de los individuos con HH menores de 30 años la enfermedad es clínicamente silente. Lo habitual es que el examen físico y las pruebas hepáticas de laboratorio sean normales. Sin embargo, en este grupo etario ya puede detectarse un aumento del nivel de la FS, la STf o ambas, con un aumento de la concentración hepática de hierro detectada por la RM T2* o por biopsia hepática.
Con la progresión de la sobrecarga férrica es frecuente observar hepatomegalia, que se detecta hasta en un 95% de los casos, acompañada usualmente de alteraciones en el metabolismo del hierro con elevación de la FS, la STf y las transaminasas, que habitualmente es leve.1
La acumulación creciente de hierro a lo largo de los años provoca injuria hepática que puede evolucionar a fibrosis, cirrosis e incluso HCC.1, 5-7
En aquellos pacientes no tratados la progresión a fibrosis avanzada (F3) o cirrosis se observa hasta en el 25% de los varones y en el 8% de las mujeres. Los factores de riesgo asociados a cirrosis son la presencia de FS > 1000 ng/ml, AST elevada, concentración hepática de hierro > 200 µmol/g, DM, artritis, depleción de hierro por sangrías > 9,6 g o ingesta alcohólica elevada. Los portadores de cirrosis tienen un riesgo incrementado de desarrollar HCC, estimado entre 12 y 200 veces superior a la población sana. Los pacientes con HH C282Y con FS ≥ 2000 ng/ml al momento del diagnóstico tienen mayor riesgo de HCC y mayor mortalidad. La incidencia de HCC a los 10 años es de aproximadamente 6% a 10%, con mayor riesgo en varones que en mujeres. Por ello, los portadores de HH con fibrosis avanzada (F3 y F4) deben ser incluidos en programas de vigilancia para detección precoz del HCC.20
Una de las causas del diagnóstico en estadios más tempranos es la investigación de sobrecarga de hierro cuando se encuentra aumento de las aminotransferasas AST o ALT (aspartato aminotranferasa y alaninoaminotransferasa), así como sobrecarga de hierro hepática en una RM de abdomen, solicitada por otros motivos. De forma similar a lo reportado en otras publicaciones, nuestro grupo encontró cirrosis en el 42,9% de los pacientes diagnosticados con HH entre 1991 y 2009 (n= 99), en contraste con el 23% de los pacientes diagnosticados entre 2009 y 2021 (n= 177), p = 0.002.
Artritis
La artritis es una manifestación clínica frecuente de la HH. Se ha descripto con una prevalencia que oscila entre 10% y 86,5%.22-23 En nuestra serie la hemos observado en el 24% de los casos. Como fue mencionado, la artritis se ha reportado entre los factores asociados a HH con fibrosis hepática avanzada.23
En una revisión reciente sobre la fisiopatogenia de la artritis en la HH, los autores concluyeron que, si bien no se conoce el mecanismo de la lesión, este sería similar a las enfermedades por depósito de pirofosfato de calcio en las articulaciones. Ambas entidades tienen un fenotipo similar, con compromiso frecuente de la segunda y tercera articulación metacarpofalángica, muñecas y hombros.
Un estudio sueco, realizado a partir de un registro nacional de datos en 3531 pacientes con HH, mostró que las artropatías no infecciosas fueron más frecuentes que en individuos sanos, así como también el reemplazo de rodilla y de cadera.24
Osteoporosis
La prevalencia de osteoporosis en HH se ha reportado entre 25% y 34% y la de osteopenia entre 40% y 79%.25-26
La osteopatía es más frecuente a nivel del cuello femoral y se correlaciona con el grado de sobrecarga de hierro.
El mecanismo fisiopatológico por el cual se produce la osteopatía en la HH aún no se comprende por completo. Se ha propuesto que la toxicidad directa del hierro puede tener un rol importante en la patogénesis. La acumulación de hierro en los huesos podría llevar a una disminución en la formación ósea y a un aumento en la reabsorción, lo que resultaría en una disminución de la densidad mineral y aumento en el riesgo de fracturas.25
Diabetes mellitus
En las últimas dos décadas se ha observado una disminución de la prevalencia de DM en pacientes con HH de alrededor de 35% a 18%, al comparar individuos con este diagnóstico antes y después de la introducción del test genético HFE en 1996. En contrapartida, la alteración de la prueba de tolerancia oral a la glucosa en estos dos periodos mostró un incremento de 6,7% a 13%, explicado por un diagnóstico más temprano de la HH.27 En los pacientes evaluados por nuestro grupo, se diagnosticó DM en 19% de los casos hasta 2009 y 11% desde esa fecha hasta 2021; no detectamos un incremento estadísticamente significativo de la prevalencia de la prueba de tolerancia oral a la glucosa alterada al comparar los periodos mencionados (datos no publicados).
En la actualidad se considera que el riesgo incrementado de DM en la HH es de causa multifactorial. Entre los factores involucrados se incluyen: el depósito de hierro en las células β de los islotes pancreáticos, la cirrosis, el antecedente de DM en familiares de primer grado, el aumento del índice de masa corporal, la resistencia a la insulina, el síndrome metabólico y el estrés oxidativo.28
Hipogonadismo
El hipogonadismo hipogonadotrófico (HHg) es una manifestación infrecuente en la HH tipo 1, con una prevalencia estimada de alrededor del 6%. Se produce por la acumulación de hierro en la glándula pituitaria y se puede observar en estadios avanzados de la HH, sobre todo en varones. En la HH tipo 2, el HHg suele ser el motivo de consulta, alcanzando una prevalencia del 98%.29
En la HH tipo 1, el HHg se asocia a cirrosis, ferritina > 1500 ng/ml y DM, por lo que es considerado un marcador de enfermedad avanzada.1, 28-29
El HHg se manifiesta clínicamente por disminución del vello corporal, disminución de la libido, disfunción eréctil, hipo o azoospermia, infertilidad, atrofia testicular de grado variable y, raramente, ginecomastia.29 En nuestros pacientes hemos observado trastornos de la libido con menor frecuencia en los últimos años; 31% hasta 2009 y 16% desde 2010 hasta 2021(p = 0.003) (datos no publicados).
El panhipopituitarismo es muy raro en la HH. Habitualmente la secreción de ACTH y TSH están preservadas.29
Disfunción tiroidea, adrenal y paratiroidea
Si bien en los portadores de HH el exceso de hierro se deposita también en la tiroides, es muy infrecuente la detección de hipo o hipertiroidismo primario.
La disfunción adrenal y paratiroidea han sido reportadas raramente.28
Manifestaciones cardiovasculares
El compromiso cardiovascular en los portadores de HH HFE se manifiesta en la fase tardía de la enfermedad, habitualmente cuando ya están afectados otros órganos.5-7, 30
Las manifestaciones clínicas cardíacas no son frecuentes y se deben al depósito de hierro en el miocardio y en el sistema de conducción, lo que da lugar a diferentes grados de hipertrofia y dilatación ventricular. Inicialmente se expresa como un trastorno de la función diastólica; posteriormente como cardiopatía dilatada e insuficiencia cardíaca congestiva.30
El electrocardiograma no es un método diagnóstico de valor en los pacientes asintomáticos o escasamente sintomáticos.1,30
La ecocardiografía bidimensional es útil en estadios avanzados de la enfermedad para detectar la disfunción diastólica y la hipertrofia, pero no en pacientes con HH en estadios precoces.30,33
La ecografía doppler bidimensional speckle tracking (Eco strain 2D) en los enfermos con HH sin sobrecarga cardíaca de hierro muestra alteraciones cuyo impacto clínico aún no ha sido definido.33 En un estudio prospectivo realizado en 23 pacientes con diagnóstico reciente de HH sin sobrecarga de hierro cardíaca evaluados con Eco strain 2D, observamos que presentaban una disminución significativa del strain global con mayor compromiso de la deformación radial y circunferencial, hallazgo que podría interpretarse como una manifestación subclínica. Estas alteraciones no se correlacionaban con los parámetros bioquímicos hepáticos ni del metabolismo del hierro ni con la presencia o ausencia del gen HFE.31-32 Resultados similares fueron reportados por otros autores, inclusive la normalización del strain con la fleboterapia.34
Manifestaciones dermatológicas
En 1977 Chevrant–Breton y col. refirieron que, de 100 pacientes con HH avanzada, un 98% presentaban hiperpigmentación de la piel. En la actualidad, entre un 5% y un 28% de los portadores presentan melanodermia.7,17,35 Esta última se produce porque el depósito de hierro estimula la síntesis de melanina.35
Cuando la hiperpigmentación es persistente y no relacionada con la exposición al sol se debe a una sobrecarga sistémica de hierro severa, que suele presentarse en individuos no tratados con enfermedad hepática avanzada, habitualmente en estadio de cirrosis.17
Infecciones
Las bacterias siderofílicas, como el Vibrio vulnificus y la Yersinia enterocolitica, pueden ocasionar infecciones severas en los portadores de sobrecarga férrica, con elevada mortalidad, mientras que en los pacientes sin exceso de hierro son moderadamente patogénicas.36
Estrés oxidativo en la HH
La sobrecarga de hierro se asocia con estrés oxidativo, que se produce por el desbalance entre un incremento de los radicales libres y un detrimento del sistema antioxidante.2-3 La coenzima Q10 (CoQ10) es un potente antioxidante endógeno y cofactor en la cadena mitocondrial de transporte de electrones.
En un estudio en portadores de HH de reciente diagnóstico no tratados, analizamos los niveles de CoQ10 y los comparamos con controles sanos. Observamos una disminución estadísticamente significativa del nivel sérico de CoQ10 y de vitamina E, independientemente de la edad, sexo, estadio de la fibrosis, severidad de la sobrecarga de hierro hepático, presencia o ausencia de genes HFE, niveles de ferritina sérica, ferremia, STf, vitaminas A y C, aminotransferasas y bilirrubina. Esta disminución de la concentración de CoQ10 podría expresar un desbalance entre el estrés oxidativo y el sistema antioxidante. La observación sugiere que en portadores de HH, una suplementación con CoQ10 podría complementar el tratamiento con flebotomías, aunque se requieren más estudios para avanzar con esta propuesta.37
Diagnóstico
El diagnóstico de HH comienza con un concepto clásico en medicina: para diagnosticar una enfermedad se debe pensar en la posibilidad de la misma al elaborar los diagnósticos presuntivos.
Ante la sospecha de HH, deben estudiarse los marcadores séricos del metabolismo del hierro: FS, Tf, Ferremia y STf. Por el momento no se recomienda investigar el nivel de HEP plasmática o urinaria, debido a que las técnicas actualmente disponibles no están estandarizadas y los resultados son muy dispares entre sí.3,6
El hallazgo de FS elevada y/o STf > 40% – 45% orienta al diagnóstico presuntivo de HH. Sin embargo, siempre se deben investigar otras etiologías y comorbilidades, debido a que entre un 58% y 70% de los pacientes con hiperferritinemia no tienen sobrecarga de hierro (Tabla 2).
Por otro lado, el hallazgo de otras patologías no excluye el diagnóstico de HH. También se debe tener presente que una STf > 50% se puede detectar cuando la Tf está disminuida, como suele observarse en las hepatopatías crónicas avanzadas de cualquier etiología.38
Tabla 2. Causas de hiperferritinemia
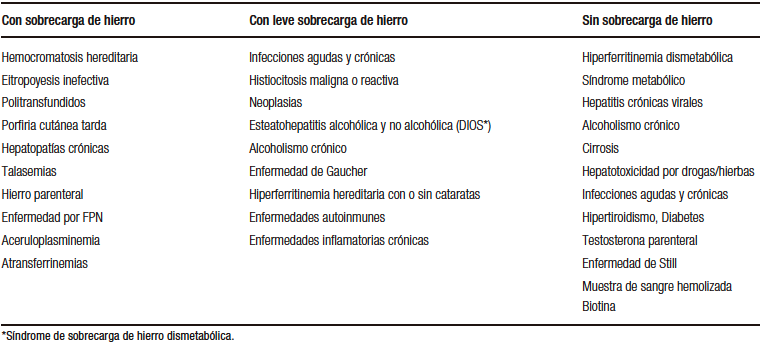
Es necesario estudiar el metabolismo del hierro en adultos que refieran antecedentes de hemocromatosis en familiares de primer grado, astenia, artropatías, osteoporosis, DM2, enfermedades hepáticas crónicas y HCC, así como también en individuos que presentan hipertransaminasemia.5-7
Cuando la FS y la STf se encuentran en niveles elevados, se debe continuar el estudio con la búsqueda de las mutaciones C282Y y H63D del gen HFE. Algunas publicaciones sugieren que solamente se debe investigar la mutación C282Y. Sin embargo, las Guías de práctica clínica sobre hemocromatosis de la EASL mencionan que: la identificación de la variante genética específica causante de la enfermedad no es necesaria ni suficiente para el diagnóstico en pacientes con hemocromatosis de inicio en la edad adulta, ya que dicho diagnóstico se basa en criterios fenotípicos.6
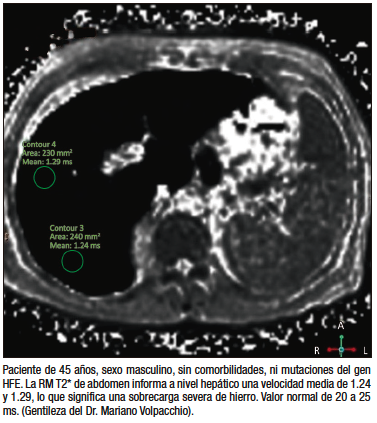
En los pacientes con FS y STf elevadas con o sin homocigosis C282Y, el estudio que se debería hacer a continuación es la RM T2*, procedimiento no invasivo de alta reproducibilidad, aunque costoso, que tiene una elevada sensibilidad y especificidad para detectar y ponderar el hierro hepático y en otros órganos (bazo, páncreas, corazón y cerebro) (Figura 4).39-40 Un aspecto controversial es la detección de sobrecarga de hierro esplénica en forma paralela a la hepática en pacientes con diagnóstico presuntivo de HH. Algunas publicaciones sugieren que solo se puede detectar en las hemocromatosis secundarias. Sin embargo, otras publicaciones no coinciden con este concepto. En nuestra experiencia, en 113 pacientes con diagnóstico bioquímico e histológico de HH a los que se les realizó RM T2*, con o sin mutaciones del gen HFE y sin otras causas que justifiquen el exceso de hierro, detectamos sobrecarga férrica esplénica en un 47% de los casos, siendo estadísticamente más frecuente en aquellos con FS > 1000 ng/ml, hepatomegalia y manifestaciones articulares. Por lo tanto, en nuestra opinión, creemos que el hallazgo conjunto de exceso de hierro hepático y esplénico no invalida el diagnóstico de HH, pero en estos casos es necesario recurrir a la biopsia hepática para confirmar el diagnóstico de HH, aún en aquellos pacientes con mutaciones HFE.41
Figura 4. Resonancia magnética T2*
Cuando la RM y los parámetros bioquímicos del metabolismo del hierro informan una sobrecarga férrica, se detecta la mutación homocigota C282Y, FS < 1000 ng/ml y el enfermo no tiene comorbilidades, se puede concluir en el diagnóstico de HH HFE5-7 y no es necesario efectuar una biopsia hepática para comenzar el tratamiento. Sin embargo, ante la ausencia de la mutación el estudio histológico es clave. De todas maneras, en presencia o ausencia de la mutación, si el enfermo tiene FS > 1000 ng/ml o hipertransaminasemia, se debe implementar el estudio histológico debido a que estos individuos tienen un riesgo de cirrosis superior al 40%. En nuestra serie, los pacientes con HH con o sin mutaciones HFE y con FS > 1000 ng/ml tienen un riesgo 5 veces mayor de ser portadores de cirrosis (datos no publicados).
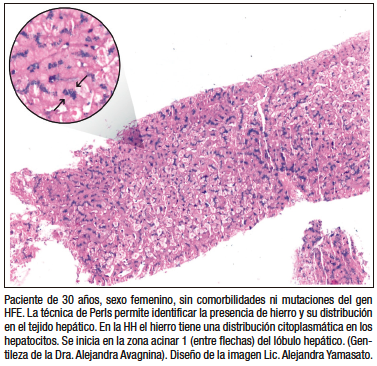
El tejido hepático obtenido por biopsia se analiza con las tinciones habituales y con la tinción de Perls para evaluar la presencia de hierro y su distribución. En la HH se observa el patrón parenquimatoso (citoplasma de los hepatocitos) con ausencia o presencia de un leve infiltrado inflamatorio y diferentes estadios de fibrosis. En la eritropoyesis ineficaz en no transfundidos la distribución del hierro hepático es similar al de la HH, por lo que este diagnóstico debe ser investigado durante la evaluación del paciente con sobrecarga de hierro (Figura 5). La cuantificación del hierro en el tejido puede hacerse en forma semicuantitativa en una escala de 1 a 4, midiendo la concentración de hierro hepático por espectrofotometría de absorción atómica en tejido fresco y parafinado, o mediante el índice de hierro.5-7, 42
Figura 5. Tinción de Perls en tejido hepático obtenido por biopsia por punción
Los métodos no invasivos para evaluar la presencia de fibrosis, como la elastografía, ARFI, los puntajes de los índices APRI y FIB-4 han sido validados para hemocromatosis en pocos estudios, por lo que por el momento no hay consenso acerca de su uso.35 La elastoresonancia, de gran utilidad para investigar fibrosis en enfermedades hepáticas, en la sobrecarga férrica hepática informa resultados difíciles de interpretar debido a que el exceso de hierro del órgano interactúa con el impulso electromagnético de la RM.
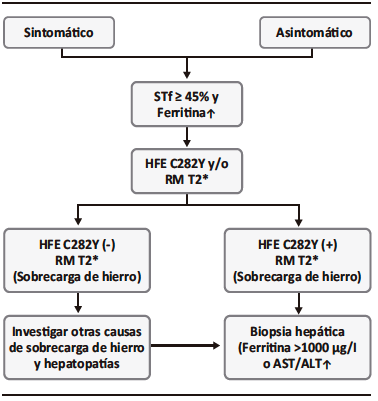
Con el objetivo de resumir el diagnóstico de estos pacientes se propone el algoritmo de la Figura 6.
Figura 6. Algoritmo diagnóstico
Tratamiento
El objetivo del tratamiento de los pacientes con HH es remover el exceso de hierro, para disminuir y evitar el daño de órganos blancos.
La reducción del exceso de hierro mejora la morbilidad y la mortalidad de estos pacientes, sobre todo cuando el tratamiento se implementa antes del desarrollo de DM y/o cirrosis. Además, podría disminuir el estadio de fibrosis en el subgrupo de pacientes que no tienen DM o edad avanzada.43 A su vez, los portadores de HH con valores de FS < 1000 ng/ml tratados tienen una tasa de mortalidad similar a la esperada en la población general.44
En la encuesta internacional realizada por la Asociación de Hemocromatosis del Reino Unido, en la que participaron 1998 pacientes con HH, luego de iniciado el tratamiento se informó mejoría de la astenia, trastornos menstruales, DM, disfunción de la glándula pituitaria, trastornos sexuales, artritis o dolores articulares, alteraciones respiratorias y/o cardiovasculares en más del 70% de los casos.17
Recientemente, en el estudio Mi-Iron (n= 104) en el que se evaluó la utilidad de la eritroaféresis en pacientes con sobrecarga de hierro moderada (Ft 300-1000 ng/ml), se observó que paralelamente al descenso de los niveles de hierro disminuían significativamente síntomas como la fatiga, asociado a una disminución de marcadores de estrés oxidativo.45
El tratamiento de elección, por su bajo costo y buena tolerancia, son las flebotomías. Comprende una etapa inicial de inducción y una posterior de mantenimiento. La inicial consiste en realizar flebotomías cada 7-14 días con el objetivo de alcanzar valores de FS entre 50 y 100 ng/ml.
Luego se continúa con una fase de mantenimiento, en la que se realizan sangrías con el objetivo de mantener los valores de FS dentro del rango previamente mencionado, para prevenir que el hierro vuelva a acumularse.5-7
En segunda instancia se ubica la eritroaféresis de grandes volúmenes, de menor disponibilidad. Este procedimiento consiste en remover selectivamente la masa de glóbulos rojos, disminuyendo el tiempo y número de procedimientos necesarios.5-7, 46
Cuando existen contraindicaciones para realizar flebotomías o eritroaféresis una opción terapéutica es el uso de quelantes. En la HH el fármaco con mayor experiencia de uso es la deferoxamina, que tiene un costo elevado y efectos adversos frecuentes. Por otra parte, la experiencia con deferasirox y deferiprona es mucho menor.5-7
En relación a la alimentación, no hay estudios que hayan demostrado que una dieta baja en hierro represente un beneficio adicional en los pacientes con HH que tienen una adherencia adecuada al tratamiento. A pesar de esto, se recomienda evitar el consumo de alimentos o suplementos que contengan hierro.5-7
Conclusiones
La HH es una enfermedad que en Sudamérica tiene una baja prevalencia de la mutación C282Y (< 85%). Sin embargo el fenotipo bioquímico-histológico, las manifestaciones clínicas, la historia natural y la respuesta al tratamiento son similares a los que presentan los portadores de HH que tienen la mutación. La incorporación de la técnica diagnóstica NGS permitirá investigar y definir los polimorfismos implicados en la región. En los adultos que refieran antecedentes de hemocromatosis en familiares de primer grado, astenia, artropatías, osteoporosis, DM, enfermedades hepáticas crónicas, HCC e hipertransaminasemia debe estudiarse el metabolismo del hierro. Cuando los pacientes tienen sobrecarga de hierro en ausencia de la mutación C282Y, se debe recurrir a la biopsia hepática para realizar el diagnóstico. El tratamiento de elección es la terapia con flebotomías o la eritroaféresis, que pueden revertir el daño producido por la sobrecarga de hierro y, a nivel hepático, retrogradar la fibrosis avanzada y la cirrosis. Aunque el riesgo de HCC disminuye, no desaparece, por lo que estos pacientes deben ser incluidos en un programa de vigilancia para detección precoz de esta neoplasia.
Propiedad intelectual. Los autores declaran que los datos, las figuras y las tablas presentes en el manuscrito son originales y se realizaron en sus instituciones pertenecientes.
Financiamiento. Los autores declaran que no hubo fuentes de financiación externas.
Conflicto de interés. Los autores declaran no tener conflictos de interés en relación con este artículo.
Aviso de derechos de autor

© 2023 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Yamasato F, Daruich J. Hemocromatosis hereditaria. Acta Gastroenterol Latinoam. 2023;53(2):113-125. https://doi.org/10.52787/agl.v53i2.303
Referencias
- Gan EK, Powel LR, Olynyk JK. Natural History and Managment of HFE-Hemochromatosis. Semin Liver Dis 2011;31:293-301.
- Wang CY, Babitt JL. Liver iron sensing and body iron homeostasis. Blood 2019;133:18-29.
- Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020;105:260-272.
- M. Wojciechowska M, Wisniewski OW, Kolodziejski P, et al. Role of Hepcidin in physiology and phatophysiology. Emerging experimental and clinical evidence. J PhysiolPharmacol 2021;
72:23-33. - Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG Clinical Guideline: Hereditary Hemochromatosis. Am J Gastroenterol 2019;114:1202-1218.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines on haemochromatosis. J Hepatol 2022;77:479-502.
- Bacon R, Adams PC, Kowdlry KV, et al. Diagnosis and Management of Hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:328-343.
- Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Hum. Genome Epidemiol. Am J Epidemiol 2001;154:193-206.
- Girelli D, Busti F, Brissot P, et al. Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood 2022;139:3018-3029.
- European Association for the Study Of The Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol 2010;53:3-22.
- Campodónico M, López MA, Fay F, et al. Premio Roberto Domecq de la Asociación Argentina para el Estudio de las Enfermedades del Hígado: “Determination and Prevalence of Hemochromatosis Genes in Patients with Hereditary Hemochromatosis”, 2000.
- Barreyro FJ, Bernard H, Elizondo K, Marucci RS. Prevalence of HFE genes C282Y, H63D, and S65C in Patients with Hemochromatosis from the Province of Misiones, Argentina. Congreso Semana Panamericana de Enfermedades Digestivas 2014, GP-110. Buenos Aires, Argentina.
- Santos PC, Cançado RD, Pereira AC, et al. Hereditary hemochromatosis: mutations in genes involved in iron homeostasis in Brazilian patients. Blood Cells Mol Dis 2011;46:302-307.
- Bismuth M, Peynaude-Debayle E. Management of HFE-related haemochromatosis. Association française pour l’étude du foie (AFEF) 2005.
- Haute Autorité de Santé, Francia. Hémochromatose liée au gène HFE (type 1), Juin 2012.
- Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron overload screening in a racially diverse population. N Engl J Med 2005;352:1769-1778.
- Smith KJ, Fife-Schaw C, Dibb B, Griffiths W. Living with the impact of iron overload: report from a large survey of people with haemochromatosis. Haemochromatosis UK 2018. https://www.haemochromatosis.org.uk/Handlers/Download.ashx?IDMF=c519f05d-d656-4ec3-8edf-7cbf39bb57d4
- Francazani AL, Piperno A, Valenti L, et al. Hemochromatosis in Italy in the Last 30 Years: Role of Genetic and Acquired Factors. Hepatology 2010;51:501-510.
- Fitzsimons EJ, Cullis JO, Thomas DW, et al.; British Society for Haematology. Diagnosis and therapy of genetic haemochromatosis (review and 2017 update). Br J Haematol 2018;181:293-303.
- Ogilvie C, Fitzsimons K, Fitzsimons EJ. Serum ferritin values in primary care: are high values overlooked? J Clin Pathol 2010;63:1124-1126.
- Richardson A, Prideaux A, Kiely PDW. Haemochromatosis: unexplained MCP or ankle arthropathy should prompt diagnostic tests; findings from two UK observational cohort studies. Scand J Rheumatol 2016;46:69-74.
- Andersson L, Powell LW, Ramm LE, et al. Arthritis prediction of advanced hepatic fibrosis in HFE hemochromatosis. Mayo Clin Proc 2022;97:1649-1655.
- Mitton‑Fitzgerald E, Gohr CM, Williams CM, Rosenthal AK. Identification of Common Pathogenic Pathways Involved in Hemochromatosis Arthritis and Calcium Pyrophosphate Deposition Disease: a Review. CurrRheumatol Rep 2022;24:40-45.
- Elmberg M, Hultcrantz R, Simard JF, et al. Increased risk of arthropathies and joint replacement surgery in patients with genetic hemochromatosis: a study of 3,531 patients and their 11,794 first-degree relatives. Arthritis Care Res (Hoboken) 2013;65:678-685.
- Yamasaki K, Hagiwara H. Excess iron inhibits osteoblast metabolism. Toxicol Lett 2009;191:211-215.
- Guggenbuhl P, Deugnier Y, Boisdet JF, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int 2005;16:1809-1814.
- Barton JM, Acton RT. Diabetes in HFE Hemochromatosis. J Diabetes Res 2017;2017:9826930.
- Pelusi C, Gasparini DI, Bianchi N, Pasqueli R. Endocrine dysfunction in hereditary hemochromatosis. J Endocrinol Invest 2016;39:837-847.
- El Osta R, Grandpre N, Monnin N, et al. Hypogonadotropic hypogonadism in men with hereditary hemochromatosis. Basic Clin Androl 2017; 27:13. DOI 10.1186/s12610-017-0057-8
- Danilowicz-Szymanowicz L, Swiatczak M, Sikorska K, et al. Pathogenesis, Diagnosis, and Clinical Implications of Hereditary Hemochromatosis-The Cardiological Point of View. Diagnostics (Basel) 2021;11:1279. DOI: 10.3390/diagnostics11071279
- Aladio JM, Yamasato F, Saad AK, et al. Two-dimensional Strain Echocardiography in the Evaluation of Ventricular Function in Patients with Newly Diagnosed Hereditary Hemochromatosis. Rev Argent Cardiol 2019;87:449-455.
- Saad AK, Aladio JM, Yamasato F, et al. Analysis of The Left Atrial Function Using Two-Dimensional Strain in Patients with Recent Diagnosis of Hereditary Hemochromatosis. Curr Probl Cardiol 2022. DOI.org/10.1016/j.cpcardiol.2021.100903
- Rozwadowska K, Daniłowicz-Szymanowicz L, Fijałkowski M, et al. Can two- dimensional speckle tracking echocardiography be useful for left ventricular assessment in the early stages of hereditary haemochromatosis? Echocardiography 2018;35:1772-81
- Byrne D, Walsh JP, Daly C, et al. Improvements in cardiac function detected using echocardiography in patients with hereditary haemochromatosis. Ir J Med Sci 2019;189:109-117.
- Chevrant-Breton J, Simon M, Bourel M, Ferrand B. Cutaneous Manifestations of Idiopathic Hemochromatosis. Arch Dermatol 1977;113:161-165.
- Khan F, Fisher MA, Khakoo RA. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J InfectDis 2007;11:482-487.
- Martinefski MR, Yamasato MF, Di Carlo MB, et al. Coenzyme Q10 deficiency in patients with hereditary hemochromatosis. Clin Res Hepatol Gastroenterol 2021;45:101624.
- Cullis JO, Fitzsimons EJ, Griffiths W, et al. Investigation and management of a raised serum ferritin. Br J Haematol 2018;181:331-340.
- Henninger B, Alustiza J, Garbowski M, Gandon Y. Practical guide to quantification of hepatic iron with MRI. EurRadiol 2020;30:383-393.
- Franca M, Marti-Bonmati L, Silva S, et al. Optimizing the management of hereditary haemochromatosis: the value of MRI R2* quantification to predict and monitor body iron stores. Br J Haematol 2018;183:491-493.
- Yamasato F, Rey E, Avagnina A, et al. Splenic iron overload in patients with hereditary hemochromatosis. XXVII Congreso ALEH, E-Poster P-19, 2022.
- Pietrangelo A, Torbenson M. Disorders of Iron Overload. MacSween´s Pathology of the Liver Ed.7º. Burt A, Ferrell L, Hubscher S Ed. Elsevier; 2018, p. 275-307.
- Prabhu A, Cargill T, Roberts N, RyanJD. Systematic review of the clinical outcomes of iron reduction in hereditary hemochromatosis. Hepatology 2020;72:1469-1482.
- Milman N, Pedersen P, á Steig T, et al. Clinically overt hereditary hemochromatosis in Denmark 1948-1985: epidemiology, factors of significance for long-term survival, and causes of death in 179 patients. Ann Hematol 2001;80:737-744.
- Bardou-Jacquet E, Morcet J, Manet G, et al. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemo- chromatosis. J Hepatol 2015;
62:682-689. - Kohan A, Niborski R, Daruich J, et al. Erythrocytapheresis with Recombinant Human Erythropoietin in Hereditary Hemochromatosis Therapy: A New Alternative. Vox Sang 2000;79:40-45.
Correspondencia: Florencia Yamasato
Correo electrónico: florenciayamasato@gmail.com
Acta Gastroenterol Latinoam 2023;53(2):113-125
