
Analia Verónica Pasqua ID· Juan Carlos Spina ID· Eduardo Mullen ID
Hospital Italiano de Buenos Aires. Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2021;51(3): 300-313
Recibido: 23/02/2021 / Aceptado: 02/08/2021 / Publicado en www.actagastro.org el 27/09/2021 / https://doi.org/10.52787/qfzz3586

Resumen
El concepto y los criterios diagnósticos de la pancreatitis autoinmune han evolucionado desde los primeros reportes de casos publicados en los años sesenta. El avance del conocimiento durante las últimas dos décadas permitió reconocer varias afecciones del páncreas, las cuales muestran, en el estudio histológico, combinaciones variables de inflamación y fibrosis. La pancreatitis autoinmune consiste, en realidad, en dos enfermedades distintas: la tipo 1 y la tipo 2. La tipo 2 es una enfermedad particular sobre la cual existe un limitado número de publicaciones y se diferencia de la “clásica” tipo 1, relacionada con la IgG-4, en cuanto a su epidemiología, las características patológicas y su historia natural. Hay pacientes con condiciones pancreáticas complejas en los cuales se demuestra un componente autoinmune y una buena respuesta al tratamiento con corticoides, pero que no reúnen los criterios sugeridos para el diagnóstico de una pancreatitis autoinmune tipo 1 o de una tipo 2. Si bien el término “pancreatitis respondedora a corticoides” ha sido usado como sinónimo de pancreatitis autoinmune tipo 1 o tipo 2, estas no son las únicas afecciones pancreáticas que mejoran con este tipo de tratamiento. En esta publicación presentamos tres casos clínicos y una revisión bibliográfica relacionada con el diagnóstico y el tratamiento de la pancreatitis autoinmune y otras condiciones menos frecuentes que presentan buena respuesta al tratamiento con corticoides.
Palabras claves. Pancreatitis autoinmune, enfermedad relacionada con IgG-4, pancreatopatías respondedoras a corticoides.
Autoimmune Pancreatitis. A Case Series and Literature Review
Summary
The concept and diagnostic criteria for autoimmune pancreatitis have evolved since the first case reports published in the 1960s. The advances in knowledge during the past two decades have resulted in the recognition of several pancreatic conditions that show, on histological evaluation, variable combinations of inflammation and fibrosis. In fact, autoimmune pancreatitis comprises two distinct diseases: type 1 and type 2. Type 2 autoimmune pancreatitis is a particular pancreatic disease about which there is limited data; it differs from the ‘classic’ IgG4-related type 1 autoimmune pancreatitis in terms of epidemiology, pathological characteristics and natural evolution. There are patients with complex pancreatic conditions where an autoimmune component and good response to steroids are demonstrated, but they do not meet the suggested criteria for the diagnosis of type 1 or type 2 autoimmune pancreatitis. Although the term steroid-responsive pancreatitis may be interpreted as a synonym to type 1 and 2 autoimmune pancreatitis, these are not the only pancreatic conditions that improve with this type of treatment. Here, we present three clinical cases and a literature review related to the diagnosis and treatment of autoimmune pancreatitis and other less frequent pancreatic disorders that present a good response to steroid therapy.
Keywords. Autoimmune pancreatitis, IgG4-related disease, steroid-responsive pancreatopathy.
Abreviaturas
AIP: Autoimmune pancreatitis.
PAI: Pancreatitis autoimmune.
ColangioRM: Colangiopancreatografía por resonancia magnética.
DWI: Secuencia de difusión.
ADC: Mapa de coeficiente de difusión aparente.
EV: Endovenoso.
FAL: Fosfatasa alcalina.
TGO: Transaminasa Glutámico Oxalacética.
TGP: Transaminasa Glutámico Pirúvica.
IgG: Inmunoglobulina G.
IgG-4: Inmunoglobulina G subclase 4.
IgE: Inmunoglobulina E.
FAN: Factor antinúcleo.
AR: Artritis reumatoidea.
ASMA: Anticuerpo anti músculo liso.
ANCA: Anticuerpo anti citoplasma de neutrófilo.
CEA: Antígeno carcinoembrionario.
H&E: Hematoxicilina y eosina.
PET: Tomografía por emisión de positrones.
TC: Tomografía computada.
DMARDs: Drogas antirreumáticas modificadoras de la enfermedad.
Introducción
La pancreatitis autoinmune (PAI) es una forma particular de pancreatitis reportada en varias regiones geográficas del mundo, aunque la mayoría de las publicaciones provienen de grupos de trabajo asiáticos (Japón), europeos (Alemania, Italia, Reino Unido) y norteamericanos. Las publicaciones provenientes de Latinoamérica son realmente limitadas, por lo cual se plantea si en esta región podrían existir formas de la enfermedad con particularidades epidemiológicas o clínicas.
La PAI se clasificó históricamente en tipo 1 y tipo 2, aunque en realidad se trata de dos entidades completamente diferentes con una característica en común: la buena respuesta al tratamiento con glucocorticoides.
Los criterios diagnósticos han ido evolucionando en el tiempo y el Consenso Internacional de la Asociación Internacional de Pancreatología ha sugerido tener en cuenta las características radiológicas (parenquimatosas y ductales), la serología, el compromiso de otros órganos, la histología y, como criterio opcional, la respuesta a corticoides.1
De acuerdo con la última versión de la clasificación de pancreatitis, basada en los factores de riesgo/etiología (TIGAR-O versión 2), la pancreatitis autoinmune puede clasificarse en tipo 1, tipo 2 e indeterminada (respondedora a corticoides no tipo 1 – no tipo 2).2 Los avances en el conocimiento de la enfermedad sugieren que, en realidad, se trata de una patología significativamente heterogénea.
En este artículo, presentamos tres casos clínicos y una revisión de la bibliografía relacionada con los aspectos diagnósticos y terapéuticos de la PAI y otras condiciones “respondedoras a corticoides” menos frecuentes.
Caso clínico 1
Paciente hombre de 48 años y nacido en Bolivia con antecedentes personales de sinusopatía, divertículos de colon, constipación e hipertrofia prostática. Sin antecedentes familiares de relevancia médica.
El paciente consultó por discomfort intermitente en hemiabdomen superior, a predominio derecho. Al interrogatorio dirigido, negó cualquier otra signo-sintomatología.
El laboratorio mostró, como datos relevantes: bilirrubina 1,65 mg/dL (directa 0,91), FAL 165 UI/l, TGO 366 UI/l, TGP 152 UI/l y amilasa 109 UI/l. La ecografía abdominal no mostró particularidades (evaluación limitada por interposición de gas intestinal).
Se realizaron resonancia magnética de abdomen con contraste EV, difusión (DWI) y colangiopancreatografía, y se observó una vesícula distendida, el páncreas aumentado de tamaño difusamente, a predominio de la cabeza, con alteración de la señal parenquimatosa en secuencias T2, con restricción en secuencias de difusión y leve menor señal a la habitual en secuencias T1, con escaso realce tras la administración del contraste EV en fases tempranas, siendo el realce progresivo hacia las fases más tardías (Figura 1). La colangioRM mostró, además, una estenosis del colédoco en su porción intrapancreática.
Figura 1

Se solicitó un perfil de autoinmunidad y marcadores tumorales, destacándose el látex AR 110,75, IgE 125,8, IgG-1 8,69, IgG-4 > 3,32 (IgG total, FAN, CA-19-9 y CEA sin particularidades).
Se efectuó una ecoendoscopía biliopancreática, en la que se observó un páncreas con tamaño aumentado y ecogenicidad disminuida difusamente; se realizó una biopsia con aguja tipo Procore de 20 Gauge en el área cefálica. Además, se observaron adenopatías peripancreáticas de aspecto inflamatorio y un engrosamiento de la pared del colédoco en su segmento intrapancreático.
El estudio histopatológico reveló un parénquima con distorsión de la histoarquitectura lobulillar-acinar, debido a un proceso inflamatorio crónico caracterizado por un infiltrado mononuclear con predominio de plasmocitos (CD 138 positivo difuso) y hasta catorce células plasmáticas IgG-4 positivas por campo de alta resolución. No se observaron células atípicas en el material examinado. Las características histológicas relevantes se muestran en la Figura 2.
Figura 2

El cuadro se interpretó como una pancreatitis autoinmune de tipo 1 y se inició tratamiento con meprednisona 40 mg matutinos por día, pantoprazol 40 mg en ayunas y trimetoprima-sulfametoxazol tres veces por semana. El paciente evolucionó favorablemente desde el punto de vista clínico, bioquímico y morfológico.
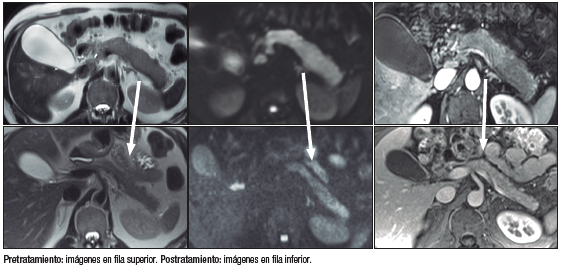
El estudio por resonancia realizado luego de cuatro semanas de tratamiento reveló un páncreas con marcada disminución de su tamaño y con persistencia de la alteración de la señal parenquimatosa hipointensa en T1 de forma difusa y del escaso realce en fase arterial, el cual se hacía más evidente en forma progresiva en los cortes más tardíos. Por otra parte, en este estudio solo se observó una sutil hiperintensidad en difusión, sin clara restricción en ADC a nivel de la cola, y disminución en número y tamaño de las estructuras ganglionares en relación con la cola del páncreas.
El paciente completó un tratamiento de inducción de la remisión con dosis decrecientes de meprednisona a partir de la quinta semana y está con dosis bajas de corticoides en un plan de tratamiento de mantenimiento.
La evolución radiológica se muestra en la Figura 3.
Figura 3. Comparación pre y postratamiento de inducción de la remisión
Desde el punto de vista funcional endocrino, el paciente presentó antes del inicio del glucocorticoide valores de glucemia compatibles con diabetes. Por otra parte, dado que evolucionó con atrofia parenquimatosa pancreática en las imágenes y presentó elastasa en materia fecal de 95 microgramos/gramo y déficit de algunos parámetros bioquímicos, como la vitamina D y el magnesio, se inició tratamiento de reemplazo con enzimas pancreáticas a dosis de 600 mg (50.000 unidades de lipasa) de pancreatina durante las comidas principales y 300 mg (25.000 unidades de lipasa) con las ingestas menores.
Caso clínico 2
Paciente mujer de 30 años, exfumadora y con cesárea un año antes de la consulta actual, refiere antecedentes familiares de adenomas de colon (vía materna).
La paciente consultó por presentar mucorrea en forma intermitente desde la cesárea y episodios de proctorragia de presentación más reciente.
Se realizó videocolonoscopía hasta íleon terminal y con limpieza óptima (Boston 9), donde se observó patología hemorroidal, áreas de mucosa congestiva intercaladas con otras de aspecto conservado desde el margen anal externo hasta la unión rectosigmoidea y mucosa congestiva con pérdida de patrón vascular y erosiones superficiales desde la unión rectosigmoidea hasta los 30 cm del margen anal externo. Se realizaron biopsias de íleon, colon derecho, izquierdo y recto, y el estudio histológico mostró un patrón de recto colitis crónica indeterminada con actividad inflamatoria aguda focal.
En contexto clínico-endoscópico, se interpretó como una enfermedad inflamatoria intestinal y se inició tratamiento con mesalazina (4 gramos vía oral + 1 gramo tópico en forma de supositorios), con buena respuesta clínica.
Un año después comenzó a presentar epigastralgia irradiada a dorso, que empeoraba con la ingesta y que no se modificaba con el tratamiento con inhibidores de la bomba de protones.
La videoendoscopía digestiva alta mostró una duodenitis congestiva y se realizaron biopsias de antro en las que no se detectó Helicobacter pylori. El laboratorio mostró, como datos relevantes: FAL 492 UI/L, TGO 245 UI/L, TGP 416 UI/L y lipasa 308 U/L.
Se realizó resonancia magnética de abdomen con contraste, difusión y colangiopancreatografía, y se observó distensión vesicular y dilatación de la vía biliar intra y extrahepática (9 mm máximo) con estenosis progresiva hacia distal. La cabeza del páncreas presentaba una formación lobulada de 48 mm, levemente heterogénea en T1 y T2, con marcada restricción en difusión y escaso refuerzo no homogéneo, pero con realce marginal tardío. Dicha formación generaba una obstrucción del conducto pancreático, cuyo contorno era ligeramente irregular en el resto de la glándula. El páncreas corporocaudal presentaba tamaño disminuido. Además, se observaron pequeños ganglios peripancreáticos cefálicos (Figura 4).
Figura 4

Se solicitaron determinaciones bioquímicas adicionales, incluyendo proteinograma, IgG total y subclases, IgE, FAN, latex AR, ASMA, ANCA C y P, CA-19-9 y CEA, sin particularidades. Glucemia normal. Elastasa en materia fecal: > 200 microgramos/gramo.
Se decidió realizar ecoendoscopía biliopancreática, la cual evidenció el páncreas hipoecoico con aumento de tamaño en cabeza y cuello (región cefálica > 7 cm), que provocaba el desplazamiento de la vía biliar; se realizó punción con aguja de 19 Gauge en la región cefálica.
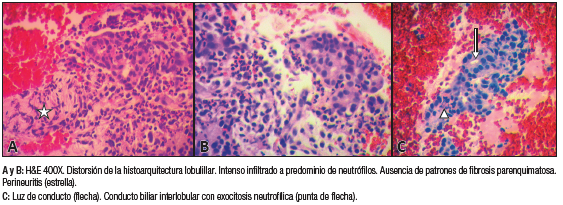
En el estudio histopatológico se observó un patrón de pancreatitis crónica con actividad inflamatoria aguda caracterizada por una notable presencia de abundantes neutrófilos. No se observaron plasmocitos IgG-4 positivos ni células epiteliales atípicas (Figura 5).
Figura 5
En contexto clínico-radiológico (y con la colopatía asociada) se interpretó como pancreatitis autoinmune de tipo 2 y se inició tratamiento con meprednisona 40 mg por día, pantoprazol 40 mg diarios y trimetoprima-sulfametoxazol tres veces por semana. La paciente evolucionó favorablemente desde el punto de vista clínico, bioquímico y radiológico en controles realizados a las cuatro semanas del inicio del tratamiento de inducción de la remisión, y al finalizar este, al cabo de doce semanas luego de una disminución gradual de la dosis de meprednisona (Figura 6).
Figura 6

No presentó evidencias de recaída después de un año de seguimiento.
Con respecto a la evolución funcional, conservó valores normales de glucemia, pero presentó una disminución de la secreción exocrina, valorada por determinación de elastasa en materia fecal, aunque con parámetros nutricionales bioquímicos conservados y ausencia de signo-sintomatología atribuible a maldigestión.
Caso clínico 3
Paciente hombre, de 47 años, cursó internación por un cuadro interpretado como pancreatitis sin patología litiásica, con trigliceridemia y calcemia normales. Refería un consumo eventual de alcohol y nunca había fumado.
No presentaba antecedentes personales de enfermedades ni medicación crónicas. No tenía antecedentes familiares de enfermedades pancreáticas. Su abuelo paterno había tenido cáncer de colon (> 80 años al momento del diagnóstico).
Inmediatamente antes de esa internación, se le habían diagnosticado un broncoespasmo y una sinusopatía tras consultar por síntomas de reciente manifestación y se le había indicado montelukast.
Se realizó estudio por resonancia, el cual mostró dos imágenes focales pancreáticas mal definidas, una localizada en la cola, de 26,8 x 20,6 mm, y otra ubicada en el sector posterior del cuerpo, de 18,2 x 13,3 mm; ambas imágenes eran hipointensas en T1 y discretamente hiperintensas en T2 y DWI, aunque sin restricción en ADC, no realzaban en fase arterial y mostraban realce progresivo en fases portal y tardía (Figura 7).
Figura 7

Se efectuó una ecoendoscopía biliopancreática y se observó una lesión levemente hipoecoica respecto del parénquima, de 22 x 21 mm, con márgenes mal definidos, negativa al doppler y sin relación con estructuras vasculares, ubicada en la cola del páncreas. Se realizó elastografía con strain ratio 1,8 (tejido blando) y punción con aguja de histología de 22 Gauge. En el cuerpo proximal del páncreas se observó otra lesión de características similares, aunque menos definida, elongada, de 10 mm. En toda la glándula se observaron estrías y focos hiperecoicos. La vía biliar y el conducto pancreático principal eran de tamaño conservado, ambos con paredes hiperecoicas.
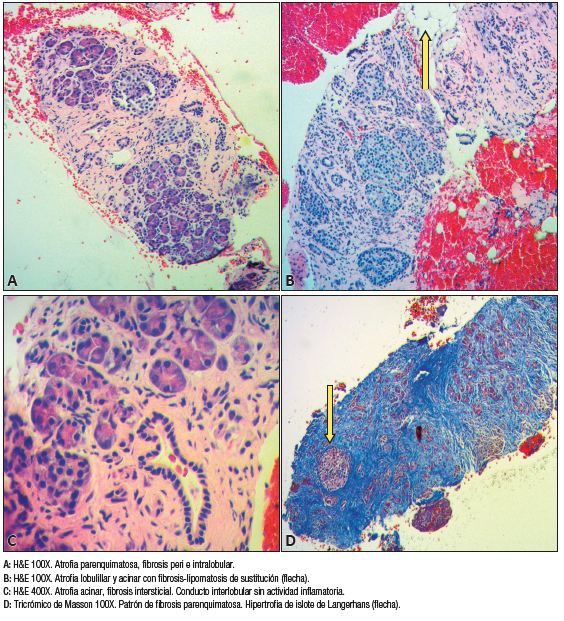
El estudio histopatológico evidenció una distorsión de la arquitectura lobulillar, atrofia lobulillar con fibrosis septal, peri e intralobulillar, hiperplasia de islotes de Langerhans con cambios reactivos de conductos pancreáticos, ausencia de plasmocitos IgG-4 positivos y ausencia de células atípicas (Figura 8).
Figura 8
En el laboratorio no presentó eosinofilia periférica. El panel de autoinmunidad (con IgG y subclases incluidas) y los marcadores tumorales solicitados (CA 19-9, CEA y cromogranina) fueron normales. La glucemia y la elastasa en materia fecal no presentaron particularidades.
Se discutió el caso en un ateneo multidisciplinario y, por impresión diagnóstica de patología inmunomediada, se decidió sugerir tratamiento con glucocorticoides, iniciando con 40 mg diarios de meprednisona.
El control con resonancia luego de cuatro semanas mostró una mejoría significativa por menor alteración de la señal y la disminución del tamaño de las dos imágenes focales pancreáticas (imagen caudal 19,6 x 15,8 mm e imagen del sector posterior del cuerpo de 14,6 mm).
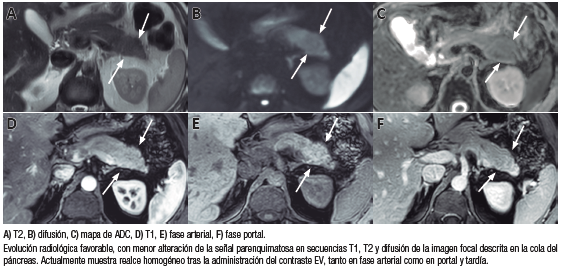
El paciente completó el esquema de inducción de la remisión de doce semanas sugerido para las pancreatitis autoinmune clásicas, y se observó la resolución, casi por completo, de las imágenes pancreáticas, con menor representación del sector distal de la cola pancreática, de aspecto atrófico y persistencia de pequeño foco hipointenso en T1, que mostró una sutil hiperintensidad en DWI, de probable origen secuelar a dicho nivel.
La evolución radiológica favorable se muestra en la Figura 9.
Figura 9
Luego de un mes de suspendido el corticoide, el paciente evolucionó con inflamación de cornetes y broncoespasmo, un cuadro clínico similar al presentado justo antes de su internación por pancreatitis.
Discusión y actualización bibliográfica
Pancreatitis autoinmune tipo 1
La pancreatitis autoinmune tipo 1 es en realidad la manifestación pancreática de la enfermedad multisistémica relacionada con la subclase 4 de la inmunoglobulina G (IgG-4), en la cual el compromiso pancreático puede ser aislado o coexistir con la afección de otros órganos, de manera sincrónica o metacrónica, de manera tal que en la atención de pacientes con esta patología es mandatorio realizar una exhaustiva revisión de los antecedentes médicos del paciente y un seguimiento de por vida de forma multidisciplinaria.
Si bien el compromiso pancreático es uno de los más representativos de la enfermedad relacionada con la IgG-4, múltiples parénquimas pueden estar afectados, entre ellos la vía biliar, los riñones, el retroperitoneo, las glándulas salivares (sialoadenitis) y las lagrimales (dacrioadenitis).
Otras manifestaciones incluyen la hipofisitis, la paquimeningitis, la tiroiditis, la enfermedad pulmonar, la pericarditis, la adenitis, la prostatitis, la mastitis y el compromiso cutáneo, entre otras.3
Teniendo en cuenta publicaciones recientes basadas en una importante cohorte de pacientes con esta patología, existen cuatro fenotipos clínicos que permitirían categorizar a la mayoría de estos enfermos en pacientes con enfermedad hepato-bilio-pancreática aislada (31%), fibrosis retroperitoneal con o sin aortitis (24%), limitada a cabeza y cuello (24%) y síndrome de Mikulicz con compromiso sistémico (22%).4 Ha sido sugerido que la fibrosis retroperitoneal y la enfermedad limitada a cabeza y cuello son más fibróticas y, por lo tanto, más refractarias al tratamiento médico.
La manifestación clínica más habitual de la pancreatitis autoinmune tipo 1 es la ictericia obstructiva indolora, aunque, con menos frecuencia, puede presentarse como una masa pancreática, un aumento difuso del tamaño del páncreas, una estenosis del conducto pancreático principal o, infrecuentemente, una pancreatitis aguda.5
Epidemiológicamente, es el tipo descrito con más frecuencia en Asia y suele afectar a individuos que cursan la séptima década de la vida, en general hombres (relación hombre: mujer de 3:1).6
Radiológicamente, el compromiso pancreático puede ser difuso, focal/segmentario o multifocal; las formas focales/segmentarias son las que plantean el diagnóstico diferencial con el adenocarcinoma ductal de páncreas, por lo cual se sugiere que el proceso diagnóstico se enfoque inicialmente en descartar esta neoplasia mediante todos los medios (incluida la punción aspirativa con aguja fina guiada por ecoendoscopía), teniendo en cuenta que este tumor es más prevalente que la pancreatitis autoinmune y que su pronóstico es especialmente desfavorable.
Si bien la tomografía computada puede sugerir el diagnóstico, en especial en la forma de presentación difusa, a nuestro criterio, la resonancia magnética de abdomen con contraste EV, difusión y colangiopancreatografía es el método por imágenes de mayor utilidad, tanto para el diagnóstico como para el seguimiento de esta patología.
Las características clásicas/típicas son el aumento del tamaño pancreático, la pérdida de lobulaciones (bordes lisos) y del patrón acinar habitual, la conservación de los planos grasos peripancreáticos y la ausencia de compromiso de estructuras vasculares. Tras la administración del contraste EV, las zonas comprometidas muestran menor realce que el parénquima sano durante la fase arterial y un realce progresivo hacia las fases más tardías. Otros elementos característicos están representados por la presencia de una marcada restricción en secuencias de difusión y la visualización de un halo peripancreático que suele ser la manifestación de una combinación de tejido fibroso e inflamatorio a dicho nivel.7
Cuando existe compromiso ductal, lo clásico es la presencia de estenosis largas (afectan > 1/3 de la longitud total del conducto pancreático principal) o múltiples con ausencia de dilatación postestenótica.
La tomografía por emisión de positrones (PET / TC) muestra la captación de fluorodeoxiglucosa, por lo cual no permite per se diferenciar una pancreatitis autoinmune de una pancreatitis crónica focal o un tumor pancreático. Este método podría ser de utilidad, en pacientes seleccionados, para valorar el compromiso en parénquimas típicamente afectados en el contexto de una enfermedad relacionada a IgG-4, tales como las glándulas salivales, los riñones y el retroperitoneo, entre otros.
Las manifestaciones hepatobiliares incluyen la colangiopatía, la colecistitis y la hepatopatía (incluido el pseudotumor inflamatorio) relacionadas a la IgG-4. La colangiopatía puede categorizarse en cuatro tipos, según el segmento del árbol biliar afectado, y se observa que el más frecuente (64%) es el tipo 1, caracterizado por la estenosis aislada del colédoco distal.8
En cuanto al estudio histopatológico, es importante señalar que es necesaria la obtención de tejido para una correcta evaluación de la arquitectura del órgano y la realización de las técnicas de inmunohistoquímica que permiten el estudio adecuado cuando se sospecha pancreatitis autoinmune.
Algunos de los rasgos histológicos que suelen observarse, en magnitud variable, en la pancreatitis autoinmune tipo 1 (también llamada “pancreatitis esclerosante linfoplasmocitaria”), son la infiltración linfoplasmocitaria periductal, la inflamación lobular, la fibrosis con una disposición “en remolinos”, la flebitis obliterativa, los folículos linfoides, > 10 plasmocitos IgG-4 (+) por campo de alta resolución (> 50 si el estudio se realiza en una pieza quirúrgica), una relación plasmocitos IgG-4 (+) / plasmocitos IgG (+) > 40% y la presencia de eosinófilos.8
Los patrones de fibrosis “en remolinos” y la venopatía obliterativa son difíciles de apreciar en el material de biopsia.
Desde el punto de vista de la histopatología, lo más dificultoso es establecer la relación IgG-4/plasmocitos IgG, dado que es técnicamente complejo correlacionar la coexpresión de ambos. Resulta práctico estimar la proporción de CD138/IgG-4 porque ambas técnicas son más específicas en la reacción y no presentan tanta dificultad de interpretación o la posibilidad de interpretaciones equívocas (especialmente en las formas no características).
Es importante tener en cuenta que en pacientes con tumores pancreáticos puede haber plasmocitos, incluso plasmocitos IgG-4 (+), a nivel peritumoral, por lo cual el esfuerzo en las biopsias de páncreas debe centrarse inicialmente en confirmar o descartar tumores.
El rol de la IgG-4 en la fisiopatología de la enfermedad no ha sido establecido con claridad. El valor de los niveles séricos de esta inmunoglobulina para el diagnóstico de esta enfermedad está definido por su especificidad (E) y sensibilidad (S), según los valores de corte considerados:
- Tab 1 Title
- Tab 2 Title
- Tab 3 Title
1,35-1,44 g/L E: 82,6% S: 87,2%
2,70-2,80 g/L E: 94,8% S: 63%
Ha sido descrito que el título suele tener correlación con la carga de la enfermedad.
Además, la relación IgG-4: IgG > 10% aumenta la especificidad diagnóstica en caso de IgG-4 normal, y los plasmablastos y células plasmáticas aumentan independientemente de la concentración de IgG-4.
Existen nuevos biomarcadores en etapa de validación, tanto para el diagnóstico como para el monitoreo de la respuesta al tratamiento y la predicción de las recaídas.4
Por otra parte, es importante tener en cuenta que los niveles de IgG-4 pueden estar elevados en la patología neoplásica, la infecciosa y en otras autoinmunes.
Otros hallazgos posibles en el laboratorio de estos pacientes son el aumento de otras subclases de la IgG, la presencia de anticuerpos inespecíficos (factor reumatoideo, anticuerpos antinucleares) a bajos títulos, la eosinofilia discreta y un aumento de la IgE.
La Liga Europea y el Colegio Americano de Reumatología han publicado recientemente una propuesta para el diagnóstico de la enfermedad relacionada con la IgG-4, la cual tiene en cuenta criterios de inclusión (compromiso clínico o radiológico característico en órganos típicamente afectados o evidencia de inflamación linfoplasmocitaria en esos órganos), criterios de exclusión (datos de alarma o que sugieran diagnósticos alternativos) y un score que, sumando ≥ 20 puntos, permitiría el diagnóstico con una especificidad del 97,8% y una sensibilidad del 82% (aun en pacientes con IgG-4 sérica normal o con una histopatología que no reúna todos los elementos característicos, teniendo en cuenta las características clínicas y/o imagenológicas, por ejemplo).9
El paciente del caso clínico 1 era un varón, cuya imagen radiológica pancreatobiliar era típica y presentaba un aumento significativo de la IgG-4 sérica y, si bien el estudio histológico de las muestras obtenidas no reveló algunas de las características clásicamente descripas en esta entidad –tales como la disposición arremolinada de la fibrosis o la venopatía obliterativa–, la suma de los datos clínicos, radiológicos, serológicos e histológicos permitió el diagnóstico de la entidad.
El tratamiento de inducción de la remisión de elección en pancreatitis autoinmune con los glucocorticoides orales y uno de los esquemas sugeridos es con prednisona a una dosis de 0,6-1,0 mg/kg/día durante cuatro semanas, con una posterior disminución gradual de la dosis diaria (tapering de a 5 mg por semana, por ejemplo) y una duración total del tratamiento de inducción de doce semanas.
Los pacientes con contraindicaciones para recibir corticoides podrían ser tratados con rituximab (anti CD20) en infusión endovenosa (dos dosis de 1 gramo separadas por dos semanas).
La necesidad de drenaje biliar por colangiografía retrógrada endoscópica debe evaluarse en cada caso particular.10
Es de buena práctica la consulta previa con infectología, para evaluar serologías, calendario de vacunación y necesidad de profilaxis antibiótica.
Con respecto a la historia natural de la enfermedad, la tasa de recaída esperada es de hasta el 60%. Las recaídas ocurren luego de discontinuar el tratamiento (67%), durante el mantenimiento (18%) o durante el tapering (15%).
Han sido descritos factores de riesgo para la recaída, los cuales son tenidos en cuenta en el momento de decidir indicar o no tratamiento de mantenimiento.
Los factores relacionados con recaídas son el compromiso pancreático difuso, la respuesta radiológica tardía o incompleta, una IgG-4 muy elevada (> x4) antes del tratamiento de inducción o persistentemente alta luego de este, el compromiso de otros parénquimas (≥ 2 órganos), la presencia de colangitis esclerosante relacionada con IgG-4 proximal y el antecedente de haber tenido una recaída previa.11
En el caso de nuestro paciente, se consideró que presentaba varios factores de riesgo para tener una recaída de la enfermedad, tales como la afección difusa del parénquima pancreático, los elevados niveles de IgG-4 sérica y la presencia de una estenosis focal en el colédoco intrapancreático con engrosamiento parietal y realce tras la administración del contraste EV, motivo por el cual se sugirió el tratamiento de mantenimiento.
Existen varios esquemas utilizados por los distintos grupos de trabajo, que incluyen a los glucocorticoides a bajas dosis (2,5-10 mg/día) o a los fármacos “ahorradores de corticoides”, entre los cuales están las drogas antirreumáticas modificadoras de la enfermedad –DMARDs– (azatioprina, metotrexate, leflunomida, micofenolato, ciclofosfamida, ciclosporina, tacrolimus, 6-mercaptopurina, iguratimod) y el rituximab. La duración de los tratamientos de mantenimiento no ha sido ampliamente consensuada y algunos grupos de trabajo han sugerido una duración de tres años.
No existe un gold standard para tratar las recaídas y las posibilidades incluyen readministrar o aumentar la dosis de corticoides ± DMARDs y rituximab.
Como se mencionó, se recomienda el seguimiento de por vida de los pacientes con enfermedad relacionada con IgG-4, ya que existen algunas evidencias de un aumento del riesgo de que se diagnostique una enfermedad maligna en ellos; esto ocurre, en general, dentro del primer año del diagnóstico de la pancreatitis autoinmune.
Es conveniente evaluar la suficiencia exocrina basal y el postratamiento mediante la determinación de elastasa I inmunorreactiva en materia fecal y una combinación de parámetros nutricionales bioquímicos, para lo cual se utilizan paneles que incluyen vitaminas liposolubles (ADEK), zinc, calcio y magnesio, entre otros.
En el caso de nuestro paciente, el nivel de elastasa < 100 µg/gramo de materia fecal junto con la hipomagnesemia e hipovitaminosis D señalaron la necesidad de indicar tratamiento de reemplazo con enzimas pancreáticas para garantizar un adecuado proceso digestivo.
Por otra parte, es importante monitorear la función pancreática endocrina. La prevalencia de diabetes en esta entidad es del 21 al 77% y el seguimiento por endocrinología para su manejo (así como para el monitoreo del eje suprarrenal y la evaluación de la salud ósea) es mandatorio.
Pancreatitis autoinmune tipo 2
Esta enfermedad es una entidad diferente a la de tipo 1 en cuanto a la epidemiología, el perfil serológico, la asociación con enfermedad en otros parénquimas, la fisiopatología, los rasgos histológicos y la evolución.
Desde un punto de vista epidemiológico, es más frecuentemente descripa en Occidente y suele afectar a individuos que cursan la quinta década de la vida y a ambos sexos por igual.6
Las manifestaciones clínicas incluyen pancreatitis aguda (≈ 50%), ictericia obstructiva indolora, masa pancreática y estenosis del conducto pancreático.5
Esta enfermedad pancreática ocurre de manera aislada o en asociación con una enfermedad inflamatoria intestinal (hasta en el 20%) como la colitis ulcerosa o la enfermedad de Crohn, la cual puede manifestarse antes, en simultáneo o después de la manifestación de la pancreatitis.
Es importante tener en cuenta el diagnóstico diferencial entre una pancreatitis autoinmune de tipo 2 y una pancreatitis por azatioprina o 6-mercaptopurina en pacientes con enfermedad inflamatoria intestinal tratados con estas drogas.
La pancreatitis autoinmune tipo 2 también se denomina “pancreatitis ducto céntrica idiopática” y la lesión epitelial granulocítica (infiltración neutrofílica del epitelio ductal y de la luz) es una característica típica; también puede haber inflamación periductal e infiltrado linfoplasmocitario, incluso con plasmocitos IgG-4 (+), aunque en concentraciones < 10 / campo de alta resolución.
La pancreatitis autoinmune tipo 2 no tiene ninguna particularidad bioquímica distintiva, pero es importante mencionar que ≈ 1/4 de los pacientes puede presentar IgG-4 sérica elevada.6
Teniendo en cuenta esta ausencia de un perfil serológico distintivo y que carece del contexto multisistémico que puede verse en la pancreatitis autoinmune tipo 1, la biopsia suele ser central para un diagnóstico definitivo.
La paciente del caso clínico 2 presentó una afección parenquimatosa focal-segmentaria del área cefálica, lo que hizo mandatoria la punción, cuyo primer y principal objetivo fue descartar el cáncer de páncreas. El intenso infiltrado inflamatorio a predominio de neutrófilos y la observación de conductos biliares interlobulares con exocitosis neutrofílica, sumado al antecedente de una enfermedad inflamatoria intestinal diagnosticada un año antes como evidencia colateral, sugirieron el diagnóstico de una pancreatitis autoinmune tipo 2.
La respuesta de la PAI tipo 2 al tratamiento con corticoides es ≈100%, pero en su historia natural la tasa de recaída esperada es de < 10%, mucho menor a la de la tipo 1, por lo cual no suele plantearse el tratamiento de mantenimiento.
En el caso de nuestra paciente, tal como ha sido descrito, la evolución clínica, la bioquímica (hepatograma) y la radiológica fueron favorables y sostenidas en el tiempo.
Pancreatitis autoinmune respondedora a corticoides no tipo 1 – no tipo 2 y otros trastornos
Las pancreatitis autoinmunes respondedoras a corticoides no tipo 1 – no tipo 2 son entidades complejas en las cuales existe un componente autoinmune, pero no reúnen los criterios para ser categorizadas como pancreatitis autoinmune tipo 1 ni tipo 2, por lo cual su diagnóstico y manejo representan un verdadero desafío.
En una reciente revisión sobre “pancreatitides” respondedoras a corticoides se hace referencia, entre otras, a las relacionadas con vasculitis/enfermedades del tejido conectivo, tales como la granulomatosis de Wegener, el lupus eritematoso sistémico o la artritis reumatoidea, aunque se menciona que la información disponible es escasa y proveniente de reportes de casos aislados o de pequeñas series.12
Por otra parte, los trastornos pancreáticos relacionados con eosinófilos incluyen, principalmente, la pancreatitis eosinofílica, la pancreatitis autoinmune y el cáncer de páncreas. La pancreatitis eosinofílica es una entidad infrecuente y se caracteriza por la infiltración del páncreas por eosinófilos de forma focal o difusa; en el proceso diagnóstico debe tenerse en cuenta que puede simular una neoplasia pancreática y que puede asociarse con gastroenteritis eosinofílica o con síndrome hipereosinofílico, dado que esto impactará en el enfoque terapéutico.13
En la práctica es relativamente frecuente observar pacientes con pancreatopatías en los cuales se sospecha un origen inmunomediado y que, luego de exhaustivas investigaciones, son tratados con corticoides y muestran una evolución favorable; en algunos de estos casos el compromiso pancreático es aislado y en otros existe un contexto sistémico que facilita la orientación diagnóstica.
El caso 3 es un ejemplo de esta situación. El paciente presentó un episodio que se interpretó como pancreatitis, sin haberse identificado una etiología biliar ni tóxico-metabólica o medicamentosa y con el antecedente de haber presentado broncoespasmo y una sinusopatía inmediatamente antes de este episodio. El estudio por resonancia mostró dos imágenes focales pancreáticas mal definidas cuyo comportamiento en algunas de las secuencias sugerían autoinmunidad. La evaluación ecoendoscópica con punción permitió, fundamentalmente, descartar una patología tumoral y en el estudio histológico no se observaron las características clásicas de la pancreatitis autoinmune tipo 1 ni de la tipo 2. El paciente tampoco presentaba particularidades en el panel de autoinmunidad en suero. Luego de un análisis minucioso y multidisciplinario de su caso, se decidió sugerir la administración de glucocorticoides por vía oral, a partir de lo cual se observó una respuesta casi completa, como fue detallado previamente.
Conclusión
Impresiona la existencia de un espectro de pancreatopatías que responden al tratamiento con corticoides, algunas con criterios diagnósticos y terapéuticos bien definidos, como las autoinmunes tipo 1 y tipo 2, y otras indeterminadas que deben manejarse de manera individualizada, personalizada, con seguimientos estrictos por equipos multidisciplinarios.
Consentimiento para la publicación. Para la confección de este manuscrito, se utilizaron datos anonimizados que no han distorsionado su significado científico.
Propiedad intelectual. Los autores declaran que los datos y las figuras presentes en el manuscrito son originales y fueron realizados en sus instituciones pertenecientes.
Aviso de derechos de autor
 © 2021 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
© 2021 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Pasqua AV, Spina JC, Mullen E. Pancreatitis autoinmune. Serie de casos y actualización bibliográfica. Acta Gastroenterol Latinoam. 2021;51(3):300-13. https://doi.org/10.52787/qfzz3586
Referencias
- Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, et al. International Consensus Diagnostic Criteria for Autoimmune Pancreatitis Guidelines of the International Association of Pancreatology. Pancreas. 2011;40:352-8.
- Whitcomb DC, for the North American Pancreatitis Study Group. Pancreatitis: TIGAR-O Version 2 Risk/Etiology Checklist With Topic Reviews, Updates, and Use Primers. Clin Transl Gastroenterol. 2019;10(6):e00027.
- Detlefsen S, Klöppel G. IgG4-related disease: with emphasis on the biopsy diagnosis of autoimmune pancreatitis and sclerosing cholangitis. Virchows Archiv. 2018;472:545-56.
- Lanzillotta M, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ. 2020;369:m1067.
- Majumder S, Takahashi N, Chari ST. Autoimmune Pancreatitis. Review. Dig Dis Sci. 2017;62:1762-9.
- Nagpal SJS, Sharma A, Chari ST. Autoimmune Pancreatitis. Review Article. Am J Gastroenterol. 2018;113:1301-9.
- Tang CSW, Sivarasan N, Griffin N. Abdominal manifestations of IgG4-related disease: a pictorial review. Insights Imaging. 2018;9(4):437-48.
- Löhr JM, Beuers U, Vujasinovic M, Alvaro D, Frøkjær JB, Buttgereit F, et al. European Guideline on IgG4-related digestive disease – UEG and SGF evidence-based recommendations. United European Gastroenterology Journal. 2020;8(6):637-66.
- Wallace ZS, Naden RP, Chari S. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol. 2020;72(1):7-19.
- Bi Y, Hart PA, Law R, Clain JE, Farnell MB, Gleeson FC, et al. Obstructive jaundice in autoimmune pancreatitis can be safely treated with corticosteroids alone without biliary stenting. Pancreatology. 2016;16:391-6.
- Okazaki K, Chari ST, Frulloni L, Lerch MM, Kamisawa T, Kawa S, et al. International consensus for the treatment of autoimmune pancreatitis. Pancreatology. 2017;17:1-6.
- Pelaez-Luna M, Soriano-Rios A, Lira-Treviño AC, Uscanga-Domínguez L. Steroid-responsive pancreatitides. World J Clin Cases. 2020;8(16):3411-30.
- Manohar M, Verma AK, Singh G, Mishr A. Eosinophilic pancreatitis: a rare or unexplored disease entity? Review paper. Gastroenterology Rev. 2020;15(1):34-8.
Correspondencia: Analia Verónica Pasqua
Correo electrónico: avpasqua@intramed.net
Acta Gastroenterol Latinoam 2021;51(3):300-313
