Edwin Jesús Pardo Escalera1 ID · Astrid Dzoara Fuentes Cruz2 ID· José Cruz Mendoza Torres3 ID
1 Departamento de Dermatología, Hospital General “Dr. Manuel Gea González”.
2 Departamento de Dermatología, Hospital General de México “Dr. Eduardo Liceaga”.
3 Instituto Nacional de Neurología y Neurocirugía “Manuel Velasco Suárez”.
Ciudad de México, México.
Acta Gastroenterol Latinoam 2024;54(2):196-201
Recibido: 21/11/2023 / Aceptado: 08/05/2024 / Publicado online el 30/06/2024 /
https://doi.org/10.52787/agl.v54i2.369

Resumen
Introducción. El liquen amiloide es el subtipo más común de amiloidosis cutánea y se caracteriza por la presencia de pápulas y placas asociadas con prurito, localizadas en pantorrillas, cara anterior de muslos e, infrecuentemente, en antebrazos. La colangitis biliar primaria es una enfermedad autoinmune del hígado caracterizada por una colangitis linfocítica destructiva y la presencia de anticuerpos específicos antimitocondriales, que condicionan lesión biliar, colestasis y fibrosis hepática progresiva. Presentación de caso. Paciente masculino de 39 años con fototipo IV, que acudió a valoración por presentar en una ocasión vómito borraceo y un episodio de evacuación melénica de escasa cantidad. Durante la exploración física, se destacó la presencia de pápulas hipercrómicas diseminadas. Se identificó un patrón colestásico con hiperbilirrubinemia a expensas de bilirrubina directa, elevación de la gamma glutamil transferasa, elevación de la fosfatasa alcalina, con enzimas hepáticas en valores normales; además, no se identificó serología positiva para hepatitis A, B y C. Los anticuerpos antimitocondriales fueron positivos por inmunofluorescencia (1:320). Se tomó una biopsia de piel en la que se observó la presencia de hiperqueratosis laminar en estrato córneo y epidermis con acantosis moderada irregular, engrosamiento de procesos interpapilares e hiperpigmentación de la capa basal. Discusión. La asociación entre amiloidosis cutánea primaria localizada y colangitis biliar primaria se ha documentado en pocas ocasiones.
Palabras claves. Colangitis biliar primaria, cirrosis hepática, amiloidosis cutánea primaria, liquen amiloide.
Generalized Amyloid Lichen Associated with Primary Biliary Cholangitis: A Case Report
Summary
Introduction. Amyloid lichen is the most common subtype of cutaneous amyloidosis and is characterized by the presence of papules and plaques associated with itching, located on the calves, anterior thighs, and rarely on the forearms. Primary biliary cholangitis is an autoimmune liver disease characterized by destructive lymphocytic cholangitis and the presence of specific antimitochondrial antibodies, which cause biliary injury, cholestasis and progressive liver fibrosis. Material and methods. Informed consent was requested from the patient for the publication of their case and accompanying photographs. Case presentation. A 39-year-old male patient with phototype IV came for evaluation due to coffee grounds vomitus on one occasion and an episode of melenic evacuation. During the physical examination, he highlighted the presence of disseminated hyperchromic papules. A cholestatic pattern was identified with hyperbilirubinemia at the expense of direct bilirubin, elevation of gamma-glutamyltransferase, elevation of alkaline phosphatase, with liver enzymes in normal values, in addition, no positive serology was identified for hepatitis A, B and C. Antimitochondrial antibodies were positive by immunofluorescence (1:320). With hemodynamic improvement, a skin biopsy was taken with the presence of laminar hyperkeratosis in the stratum corneum and epidermis with moderate irregular acanthosis, thickening of the interpapillary processes,and hyperpigmentation of the basal layer. Discussion. The association between localized primary cutaneous amyloidosis and primary biliary cholangitis has been documented rarely.
Keywords. Primary biliary cholangitis, liver cirrhosis, primary cutaneous amyloidosis, lichen amyloidosis.
Introducción
La amiloidosis cutánea primaria localizada consta de un grupo raro de manifestaciones dermatológicas causadas por el depósito extracelular de proteína amiloide en la piel previamente sana, sin compromiso de otros órganos.1 Se clasifica en liquen amiloide, amiloidosis macular, amiloidosis nodular y amiloidosis bifásica.2
El 57% de los casos de amiloidosis primaria cutánea son dados por el liquen amiloide, y se encuentra una mayor prevalencia en Asia, Sudamérica y en medio este, predominantemente en población con un fototipo III-IV.3
El liquen amiloide es el subtipo más común de amiloidosis cutánea y se caracteriza por la presencia de pápulas y placas asociadas con prurito, localizadas en pantorrillas, cara anterior de muslos y en antebrazos.3
Las lesiones características son pápulas hiperqueratósicas, hiperpigmentadas que pueden confluir y formar placas con distribución a nivel de la región pretibial; de forma infrecuente se observa un patrón generalizado.4
La colangitis biliar primaria es una enfermedad autoinmune del hígado caracterizada por una colangitis linfocítica destructiva y la presencia de anticuerpos específicos antimitocondriales, que condicionan lesión biliar, colestasis y fibrosis hepática progresiva.5
A nivel mundial la prevalencia de la colangitis biliar primaria es de 2-40 casos por cada 100.000 habitantes, con mayor predominio en mujeres, que representan el 92% de los casos, y la razón mujeres: hombres es de 10:1.6
Caso clínico
Paciente masculino de 39 años con fototipo IV, con antecedente de discapacidad intelectual moderada secundaria a trauma obstétrico, diabetes mellitus tipo 2, hipertensión arterial sistémica y enfermedad hepática no especificada en seguimiento por su médico particular. Además, antecedente de colecistectomía laparoscópica hace diez años por colecistitis aguda litiásica, y prurito generalizado desde hace diez años tratado en una sola ocasión con fototerapia no especificada.
Acudió a valoración por presentar en una ocasión vómito borráceo y un episodio de evacuación melénica de escasa cantidad. Al momento de la evaluación, presentaba hipotensión arterial (90/40 mmHg), taquicardia (110 latidos por minuto) y taquipnea (23 respiraciones por minuto).
En la exploración física se identificó una tendencia a la inatención, ictericia conjuntival, mucosa oral pálida y seca con escasos restos hemáticos, abdomen blando y doloroso a la palpación superficial y profunda a nivel del mesogastrio, hepatomegalia de 8 centímetros debajo del reborde costal derecho, sin presencia de circulación colateral superficial, y tacto rectal con presencia de materia fecal oscura a la salida del guante explorador.
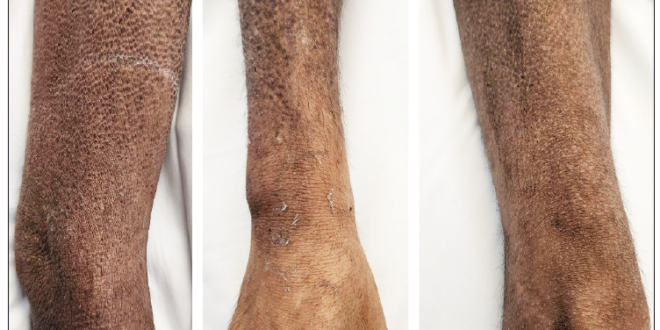
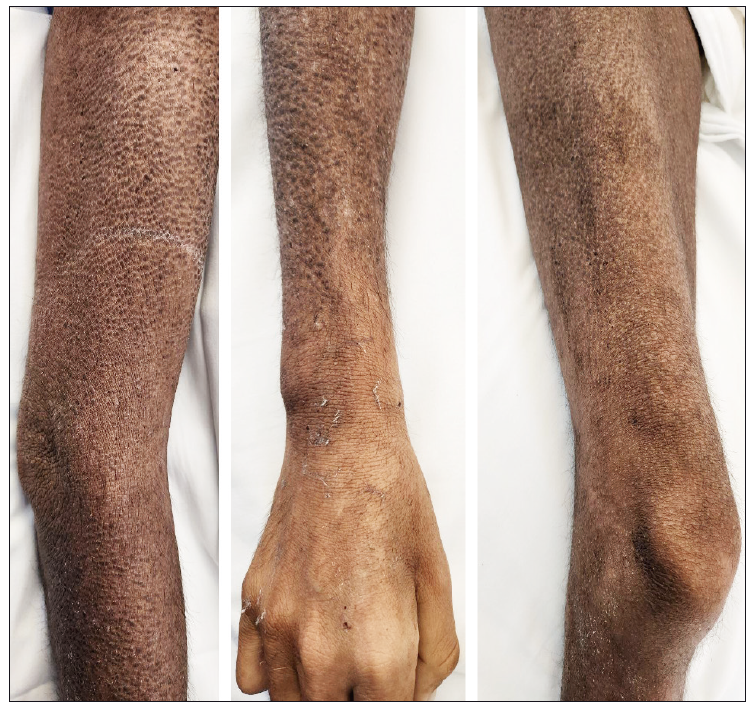
Durante la exploración física, se destacó la presencia de pápulas hipercrómicas diseminadas que llegaban a formar placas con liquenificación y costras hemáticas lineales asociadas, ubicadas en abdomen, cara anterior del tórax, espalda, región posterior de las extremidades superiores y en ambas caras de las extremidades inferiores (Figura 1).
Figura 1. Lesiones papulares hiperpigmentadas con liquenificación asociada en brazo, antebrazo y muslo derechos, respectivamente
Se inició reanimación hídrica y se transfundieron dos concentrados eritrocitarios al presentar datos de hipotensión y cifras de hemoglobina de 5,10 mg/dl, resultando en una mejoría hemodinámica y recuperación de las cifras de hemoglobina. Además, se agregaron omeprazol y octreotide.
Ante la sospecha de hemorragia digestiva, se realizó panendoscopía con sedación, la cual reportó varices esofágicas grandes, según la clasificacion de Baveno, y gastropatía portal hipertensiva. Se realizó una ligadura de varices.
Mediante los estudios de laboratorio, se identificó un patrón colestásico con hiperbilirrubinemia (3,96 mg/dL)
a expensas de bilirrubina directa (3,09 mg/dl), elevación de la gamma glutamil transferasa (366 U/L), elevación de la fosfatasa alcalina (543 U/L), con enzimas hepáticas en valores normales, aspartato aminotransferasa
(29 U/L) y alanina aminotransferasa (31 U/L); además, no se identificó serología positiva para hepatitis A, B y C. Los anticuerpos antimitocondriales fueron positivos por inmunofluorescencia (1:320), y la medición de anticuerpos antinucleares y de anticuerpos antimúsculo liso fueron negativas.
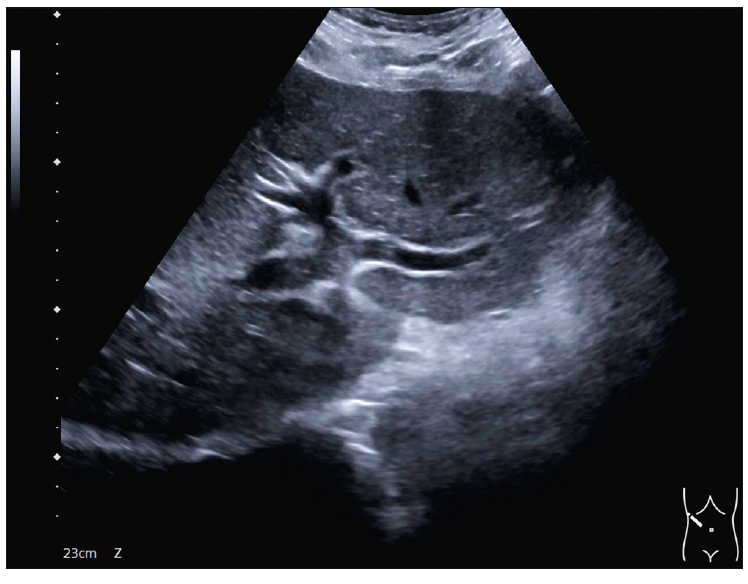
El ultrasonido de hígado mostró un aspecto micronodular heterogéneo, bordes irregulares con datos de hipertensión portal, esplenomegalia y derivaciones porto sistémicas esplenorrenales (Figura 2).
Figura 2. Ultrasonido de hígado y vía biliar que muestra datos de hepatopatía crónica e hipertensión portal
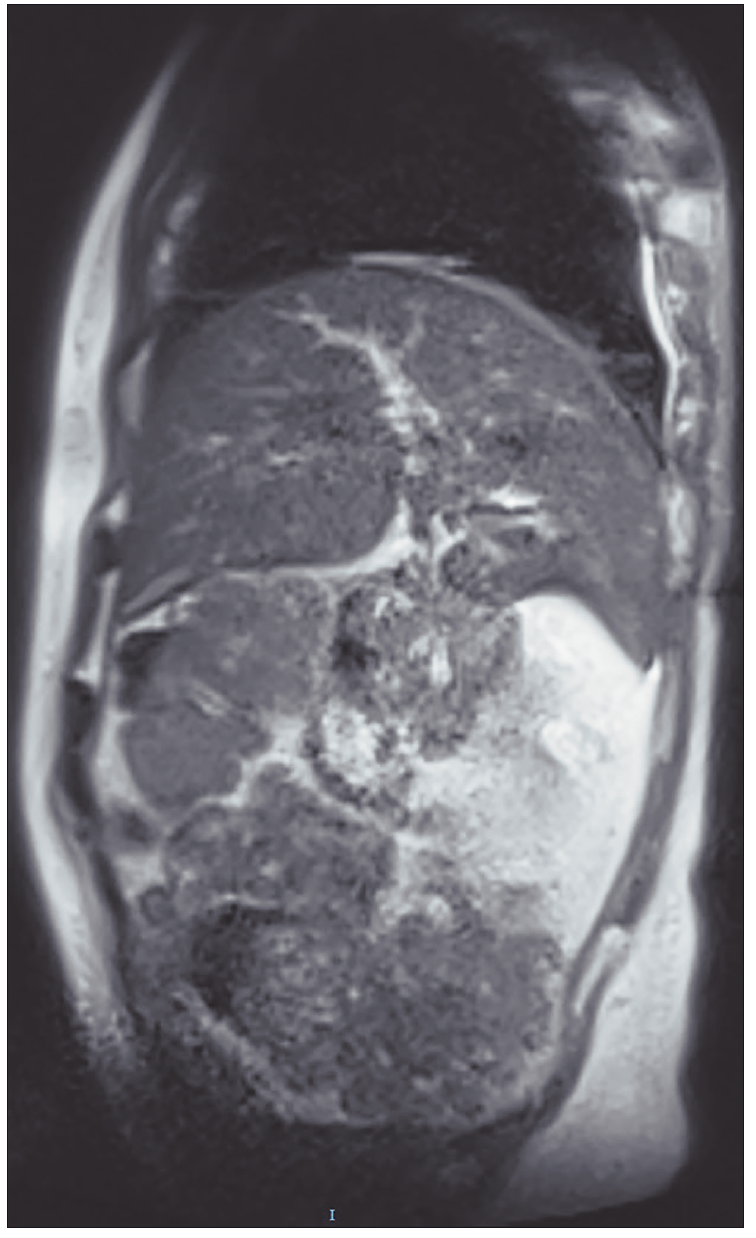
La colangioresonancia magnética identificó dilatación de la vía biliar de primer y segundo orden, vena porta de 17 mm, colaterales esplenorrenales y gástricas cortas, así como esplenomegalia (Figura 3).
Figura 3. Colangioresonancia que identifica dilatación de los conductos biliares primarios y secundarios
Al contar con mejoría hemodinámica, se tomó una biopsia de piel en la que se observó la presencia de hiperqueratosis laminar en estrato córneo y epidermis con acantosis moderada irregular, engrosamiento de los procesos interpapilares e hiperpigmentación de la capa basal. En la dermis papilar se identificaron depósitos de material eosinofílico ante el uso de cristal violeta, incontinencia pigmentaria y escasos melanófagos (Figura 4).
Además, se observó un infiltrado linfohistiocitario perivascular en la dermis reticular superficial.
Figura 4. Biopsia de piel con presencia de material eosinofílico a nivel de la dermis papilar correspondiente a amiloide. Tinción de hematoxilina y eosina
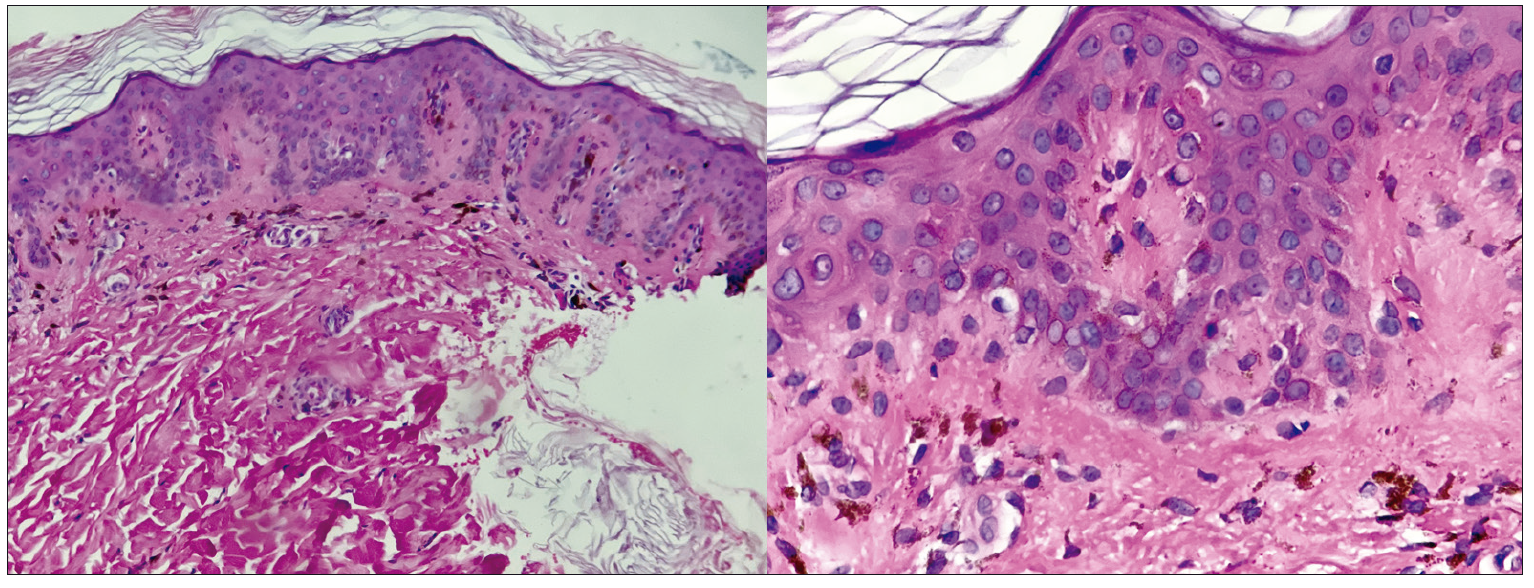
Ante la sospecha de amiloidosis sistémica con compromiso hepático, se realizó una biopsia transcutánea hepática, con reporte de colangitis destructiva no supurativa, ductopenia y cirrosis biliar con nódulos regenerativos, por lo que fue clasificado en un estadio 4 de Ludwing.
El manejo consistió en la administración de lactulosa 10 ml cada 24 horas, ácido ursodesoxicólico 300 mg cada 12 horas, colestiramina y prednisona, con mejoría del prurito.
Discusión
En la amiloidosis primaria cutánea la frecuencia de pacientes afectados con fototipo IV se ha estimado en el 29,7% de los casos, y solo el 2,7% de los casos se presentan con un patrón generalizado.7
La fisiopatología de la enfermedad sigue siendo desconocida. Sin embargo, la causa del depósito de amiloide extracelular se deriva de la necrosis de queratinocitos epidérmicos basales.8
Además, se cree que alteraciones en la señalización de la vía del receptor de oncostatina M de tipo II y del receptor de la interleucina 31 permiten la apoptosis de queratinocitos, con el subsecuente depósito de amiloide a nivel de la dermis papilar y la presencia de prurito crónico que permite un ciclo de rascado-necrosis de queratinocitos-depósito de amiloide.9
A nivel histopatológico, en el liquen amiloide se observa depósito de material globular amorfo eosinofílico en la dermis papilar, pudiendo asociarse con un incremento de la pigmentación de la capa basal, y es frecuente la presencia de acantosis e hiperqueratosis asociadas al rascado crónico.1 Estas mismas características fueron evidenciadas en nuestro paciente.
El uso de rojo congo, violeta cristal o el uso de inmunohistoquímica con citoqueratina 5/6 (CK 5/6) permiten una mejor visualización de los depósitos de amiloide en la dermis.2
El prurito en la amiloidosis cutánea primaria se cree que se produce por mutaciones que llevan a alteraciones de señalización del receptor de interleucina 31.10 Además, es el hallazgo clínico más frecuente en liquen amiloide, presentándose en el 75% de los casos.11
Por otro lado, el prurito en la colangitis biliar primaria se debe a los niveles aumentados de ácidos biliares que se acumulan en la piel y que, mediante el receptor biliar acoplado a proteína G (TGR5), media la neurotransmisión del proceso colestásico a la piel, lo que conduce al rascado crónico, la excoriación y el sangrado. Además, el depósito de melanina en la piel ocurre en la mitad de los casos, lo que lleva a la hiperpigmentación.12
El 60% de los pacientes se presentan de forma asintomática; sin embargo, los síntomas pueden aparecer entre los 2 y 4 años del diagnóstico o, incluso, presentarse cuando la enfermedad está avanzada y se evidencia cirrosis hepática.13
Las manifestaciones clínicas más comunes en la colangitis biliar primaria son la fatiga y el prurito, presentes en el 80% de los casos.14
Las pruebas de la función hepática muestran una elevación de la bilirrubina, acompañada de una elevación tanto de las enzimas hepáticas como de la fosfatasa alcalina y la gamma glutamil transferasa.15
El diagnóstico se realiza cuando existen dos de los tres criterios siguientes: 1) elevación persistente de la fosfatasa alcalina por 6 o más meses, 2) presencia de anticuerpos antinucleares o anticuerpos antimitocondriales, o 3) evidencia histológica de colangitis obstructiva no supurativa con compromiso de los conductos biliares interlobulares.16
A nivel histológico, se observan lesiones que involucran los ductos interlobulares y septales.17 Los criterios de estadificación histológica de Ludwing13 son:
Estadio 1: inflamación portal con lesión ductal.
Estadio 2: proliferación ductal y hepatitis de interfase, caracterizada por necrosis celular y apoptosis desde los tractos portales a través del parénquima hepático.
Estadio 3: disminución de la inflamación, fibrosis septal, necrosis o fibrosis.
Estadio 4: cirrosis con nódulos regenerativos.
La primera línea de tratamiento consiste en el uso de ácido ursodesoxicólico, que permite la mejora del flujo biliar a través de los ductos biliares, disminuye el daño a las células epiteliales de los ductos biliares causado por colestasis y a los hepatocitos, además de enlentecer la progresión de la enfermedad al disminuir el ataque inmunomediado a los conductos biliares intrahepáticos.18
La terapia que se dirige a disminuir la presencia y severidad del prurito se da mediante el uso de colestiramina, la cual disminuye los niveles de colesterol y metaboliza el exceso de bilis en la sangre, mejorando el prurito en el 90% de los pacientes.19
Sin la identificación y la pronta intervención, un gran número de pacientes con colangitis biliar primaria progresan hacia la insuficiencia hepática, el trasplante hepático o incluso hacia la muerte a los diez años de iniciado el padecimiento.20
Hasta el 30% de los pacientes con colangitis biliar primaria pueden presentar una forma severa y progresiva de la enfermedad, resultando en un desarrollo temprano de fibrosis hepática e insuficiencia hepática.21
La asociación entre amiloidosis cutánea primaria localizada y colangitis biliar primaria se ha documentado en pocas ocasiones; la primera vez, en una mujer de 41 años con presencia de máculas hiperpigmentadas con compromiso del tronco y piernas, quien se vio afectada por amiloidosis macular y, posteriormente, desarrolló colangitis biliar primaria con alta sospecha de amiloidosis hepática.22
Más adelante, se reportó la existencia de amiloidosis macular en brazos y piernas en un hombre de 36 años, al que se le realizó una biopsia hepática ante la sospecha de amiloidosis hepática, sin que se identificaran depósitos hepáticos de amiloide, pero se le diagnosticó una colangitis biliar primaria.23
El último caso reportado fue en una mujer de 59 años, a la que se le diagnosticó síndrome de solapamiento de hepatitis autoinmune-colangitis biliar primaria avanzada con cirrosis, síndrome de Sjögren y amiloidosis macular.24
Conclusión
En pacientes con amiloidosis cutánea se debe buscar afectación a nivel hepático, con la finalidad de identificar amiloidosis a este nivel. Sin embargo, también es de importancia identificar su asociación con hepatopatías autoinmunes. En los casos de amiloidosis cutánea y colangitis biliar primaria, es indispensable un diagnóstico oportuno para así mejorar la calidad de vida de los pacientes.
Consentimiento para la publicación. Se obtuvo el consentimiento informado por escrito del paciente o su padre, tutor o familiar, para la publicación de los datos y/o imágenes clínicas en beneficio de la ciencia. La copia del formulario de consentimiento se encuentra disponible para los editores de esta revista.
Propiedad intelectual. El autor declara que los datos y las figuras presentes en el manuscrito son originales y se realizaron en sus instituciones pertenecientes.
Financiamiento. El autor declara que no hubo fuentes de financiación externas.
Conflictos de interés. El autor declara no tener conflictos de interés en relación con este artículo.
Aviso de derechos de autor

© 2024 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Pardo Escalera E J, Fuentes Cruz A D y Mendoza Torres J C. Liquen amiloide generalizado asociado a colangitis biliar primaria: reporte de caso. Acta Gastroenterol Latinoam. 2024;54(2):196-201. https://doi.org/10.52787/agl.v54i2.369
Referencias
- Hamie L, Haddad I, Nasser N, Kurban M, Abbas O. Primary Localized Cutaneous Amyloidosis of Keratinocyte Origin: An Update with Emphasis on Atypical Clinical Variants. Am J Clin Dermatol. 2021;22(5):667-80.
- Kaltoft B, Schmidt G, Lauritzen AF, Gimsing P. Primary localized cutaneous amyloidosis-a systematic review. Dan Med J. 2013;60(11):A4727.
- Aung T, Noakes R, Murrell DF, Daniel BS, Kelati A. Primary cutaneous amyloidosis: A review of the available studies and gaps in data. Australas J Dermatol. 2023;64(2):e121-e124.
- Tursen U, Kaya TI, Dusmez D, Ikizoglu G. Case of generalized lichen amyloidosis. Int J Dermatol. 2003;42(8):649-51.
- Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17(2):93-110.
- Joshita S, Umemura T, Tanaka E, Ota M. Genetic Contribution to the Pathogenesis of Primary Biliary Cholangitis. J Immunol Res. 2017;2017:3073504.
- Guillet C, Steinmann S, Maul JT, Kolm I. Primary Localized Cutaneous Amyloidosis: A Retrospective Study of an Uncommon Skin Disease in the Largest Tertiary Care Center in Switzerland. Dermatology. 2022;238(3):579-86.
- Breathnach SM. “Amyloid and amyloidosis”. J Am Acad Dermatol. 1988;18(1):1-16.
- Tanaka A, Arita K, Lai-Cheong JE, Palisson F, Hide M, McGrath JA. New insight into mechanisms of pruritus from molecular studies on familial primary localized cutaneous amyloidosis. Br J Dermatol. 2009;161(6):1217-24.
- Lin MW, Lee DD, Liu TT, Lin FY, Chen YS, Huang CC, et al. Novel IL31RA gene mutation and ancestral OSMR mutant allele in familial primary cutaneous amyloidosis. Eur J Hum Genet. 2010;18(1):26-32.
- Mehrotra K, Dewan R, Kumar JV, Dewan A. Primary Cutaneous Amyloidosis: A Clinical, Histopathological and Immunofluorescence Study. J Clin Diagn Res. 2017;11(8):WC01-WC05.
- Chuang YY, Lee DD, Lin CS, Chang JY, Tanaka M, Chang TY, et al. Characteristic dermoscopic features of primary cutaneous amyloidosis: a study of 35 cases. Br J Dermatol. 2012;
167(3):548-54. - Carey EJ, Levy C. Primary Biliary Cholangitis: A New Era. Clin Liver Dis. 2018;22(3):xiii-xiv.
- Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386(10003):1565-75.
- Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172.
- Tan D, Goodman ZD. Liver Biopsy in Primary Biliary Cholangitis: Indications and Interpretation. Clin Liver Dis. 2018;22 (3):579-88.
- Goel A, Kim WR. Natural History of Primary Biliary Cholangitis in the Ursodeoxycholic Acid Era: Role of Scoring Systems. Clin Liver Dis. 2018;22(3):563-78.
- Trivedi HD, Lizaola B, Tapper EB, Bonder A. Management of Pruritus in Primary Biliary Cholangitis: A Narrative Review. Am J Med. 2017;130(6):744.e1-744.e7.
- Prince MI, Ducker SJ, James OF. Case-control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut. 2010;59(4):508-12.
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bersaga VN, Heathcote JE, et al. Primary biliary cirrhosis. Hepatology. 2009;50(1):291-308.
- Tafarel JR, Lemos LB, Oliveira PM, Lanzoni VP, Ferraz ML. Cutaneous amyloidosis associated with primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2007;19(7):603-5.
- González-Moreno EI, Cámara-Lemarroy CR, Borjas-Almaguer DO, Martínez-Cabriales AS, Paz-Delgadillo J, Gutiérrez-Udave R, et al. Cutaneous amyloidosis associated with autoimmune hepatitis-primary biliary cirrhosis overlap syndrome. Ann Hepatol. 2015;14(3):416-9.
- Yan X, Jin J. Primary cutaneous amyloidosis associated with autoimmune hepatitis-primary biliary cirrhosis overlap syndrome and Sjögren syndrome: A case report. Medicine (Baltimore). 2018;97(8):e0004.
Correspondencia: José Cruz Mendoza Torres
Correo electrónico: josmentorr@gmail.com
Acta Gastroenterol Latinoam 2024;54(2):196-201
