Fabian Salgueiro ID· Carol Lezama Elecharri ID· Marcela Galoppo ID· Micaela Wisniacki ID· María Solaegui ID· Alejandra Pedreira ID· Sabrina Torres ID· Elena De Matteo ID· Miguel Palmeiro ID· Pablo Volonté ID· Cristina Galoppo ID
Hospital de Niños “Ricardo Gutiérrez”.
Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2022;52(3):388-394
Recibido: 29/07/2022 / Aceptado: 06/09/2022 / Publicado online el 29/09/2022 / https://doi.org/10.52787/agl.v52i3.230

Resumen
La derivación biliar parcial externa en pacientes con colestasis intrahepática familiar progresiva y síndrome de Alagille reduce los niveles sanguíneos de sales biliares modificando su circulación enterohepática. Si esta cirugía es realizada antes de la instalación de la cirrosis, podría mejorar la sintomatología y modificar la progresión de la fibrosis hepática y, con ello, la evolución de la enfermedad, evitando o retrasando la necesidad de trasplante hepático. Presentamos los casos de ocho pacientes a quienes se les realizó una derivación biliar parcial externa: cinco con diagnóstico de colestasis intrahepática familiar progresiva, y tres con síndrome de Alagille y prurito invalidante. En todos ellos se descartó por medio de biopsia hepática la presencia de cirrosis. De los cinco pacientes con colestasis intrahepática familiar progresiva, dos se encuentran vivos, con su hígado nativo y escasa sintomatología a cuatro y veinte años del procedimiento, respectivamente. Los tres pacientes con síndrome de Alagille presentaron una mejoría de la sintomatología, con resolución en dos de ellos.
Palabras claves. Colestasis intrahepática familiar progresiva, síndrome de Alagille, derivación biliar externa.
Partial External Biliary Diversion in Progressive Familial Intrahepatic Cholestasis and Alagille Syndrome: A Case Series
Summary
Partial external biliary diversion in patients with progressive familial intrahepatic cholestasis and Alagille syndrome reduces plasma bile salt levels by modifying their enterohepatic circulation. When this surgical technique is performed before the onset of cirrhosis, the symptoms may improve. It can also modify hepatic fibrosis evolution and, thus, disease progression by avoiding or delaying liver transplantation. We present eight patients undergoing partial external biliary diversion: five patients were diagnosed with progressive familial intrahepatic cholestasis, and three of them with Alagille syndrome and severe pruritus. Presence of cirrhosis was ruled out in all cases after liver biopsy was performed. Two out of five patients with partial external biliary diversion are alive with their native liver and are almost asymptomatic four and twenty years after the procedure respectively. The three patients with Alagille syndrome improved their symptoms; two of them had complete symptom resolution.
Keywords. Progressive familial intrahepatic cholestasis, Alagille syndrome; external biliary diversion.
Abreviaturas
PFIC: Progressive Familial Intrahepatic Cholestasis.
SA: Síndrome de Alagille.
PEBD: Partial External Biliary Diversion.
DBPE: Derivación biliar parcial externa.
TH: Trasplante Hepático.
Introducción
La colestasis intrahepática familiar progresiva (PFIC, por sus siglas en inglés) y el síndrome de Alagille (SA) forman parte de las colestasis genéticas, representando aproximadamente el 25% de estas.1 La PFIC constituye un grupo de enfermedades de herencia autosómica recesiva, causado por mutaciones en los genes codificantes de proteínas de transporte canalicular.
Fisiopatológicamente, la PFIC1 obedece a la mutación de la proteína FIC1, con función de translocasa de aminofosfolípidos, que mantiene la integridad de la membrana plasmática y el normal funcionamiento de las proteínas insertas en ella, entre las cuales se encuentra la bomba exportadora de sales biliares (BSEP, por sus siglas en inglés).2 En la PFIC2 se encuentra alterada la excreción canalicular de ácidos biliares por alteraciones en la proteína BSEP. Como consecuencia, se produce su acumulación dentro del hepatocito, generando daño celular y evolución a la cirrosis.3 En estas dos patologías, la principal manifestación es la colestasis, que se presenta en los primeros meses de vida y está acompañada de prurito de difícil manejo. Con frecuencia, de no mediar tratamiento alguno, la PFIC evoluciona a enfermedad hepática terminal. En el caso de la PFIC 1 y la 2, esto ocurre dentro de los primeros años de vida, representando en la actualidad entre el 10% y el 15% de las causas de trasplante hepático (TH) en la edad pediátrica.3
El SA es una patología multisistémica de herencia autosómica dominante, causada por mutaciones en el gen JAGGED1 y, en menor medida, en el gen NOTCH2, que involucran vías de señalización celular. La principal forma de manifestación es la colestasis neonatal, si bien existe una amplia variedad de presentaciones clínicas que involucran a múltiples órganos, y estas varían desde formas subclínicas el severo compromiso de órganos y sistemas. Como consecuencia de la colestasis, algunos pacientes presentan un prurito severo y de difícil control, que suele iniciarse entre los 3 y los 5 meses de vida, aun en ausencia de ictericia. En el laboratorio, el patrón de colestasis se acompaña de hipertrigliceridemia e hipercolesterolemia, siendo esto último la causa de los xantomas que aparecen en el rostro y superficies extensoras. Respecto del compromiso hepático, entre un 10% y un 20% de los pacientes desarrollan cirrosis y enfermedad hepática terminal;4 sin embargo, el prurito y los xantomas desfigurantes pueden constituir indicaciones de trasplante hepático, aun con función de síntesis conservada.
En la actualidad no existen tratamientos curativos para las patologías descritas. En ambos casos, el tratamiento médico tiene como objetivo el abordaje nutricional, y el manejo de las complicaciones de la cirrosis y del prurito. Este último es considerado un síntoma cardinal en ambas patologías y requiere un abordaje multidisciplinario, con esquemas de tratamiento multimodales, ya que su severidad interfiere en la calidad de vida de los pacientes, dado que genera lesiones en la piel, dificultades en el sueño, irritabilidad y déficit de atención.4
La derivación biliar parcial externa (DBPE) consiste en la realización de una fístula bilio cutánea que disminuye la recirculación enterohepática de sales biliares y representa una alternativa terapéutica para estos pacientes.
Presentamos los resultados obtenidos con esta técnica quirúrgica en cuanto a las complicaciones quirúrgicas, la evolución de la enfermedad y la modificación del prurito, utilizando la escala de Whittington, en una serie de pacientes con diagnóstico de PFIC1, PFIC2 y SA.
Casos clínicos
Entre los años 2000 y 2018 se realizaron un total de 8 DBPE. En cinco pacientes con PFIC se ofreció la cirugía al diagnóstico como terapéutica de elección, mientras que en tres niños con SA la indicación de DBPE se realizó debido al prurito intenso e inmanejable con el tratamiento médico (categorizado en la escala como 3 o 4). En todos los casos se descartó la presencia de cirrosis por medio de biopsia hepática y se categorizó la fibrosis en leve, moderada o severa.
Se recabaron datos de laboratorio previos al procedimiento y un mes posterior a este, así como el último control realizado. Se registró el grado de prurito resultante de lo manifestado por el paciente, su familia y la valoración médica, y se categorizó utilizando la escala de Whittington (Tabla 1).
Tabla 1. Escala de Whittington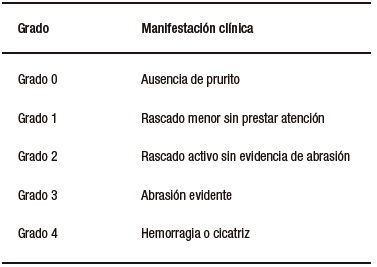
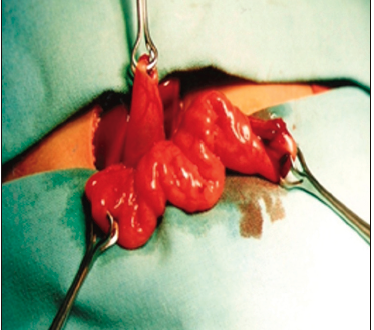
Con respecto a la técnica quirúrgica, se realizó el abordaje abdominal mediante una incisión subcostal derecha, para luego aislar un segmento de yeyuno de 10 cm de longitud, preservando su irrigación, aproximadamente a 30 cm del ángulo de Treitz (Figura 1). Posteriormente, se realizó una anastomosis entero-entérica para reconstruir el tránsito intestinal. El segmento aislado y pediculizado se anastomosó en forma isoperistáltica por un extremo al fondo vesicular (Figura 2) y, con el otro, se confeccionó una ostomía terminal exteriorizada en la fosa ilíaca derecha (Figuras 3 y 4).
Figura 1. Vesícula biliar y asa de yeyuno aislada para la derivación
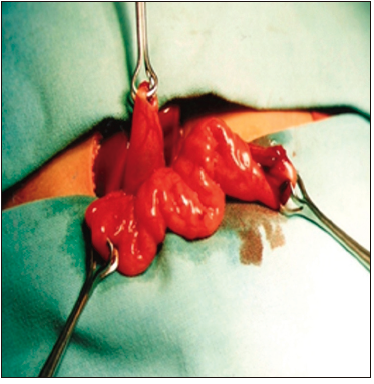
Figura 2. Anastomosis del asa yeyunal al fondo de la vesícula biliar
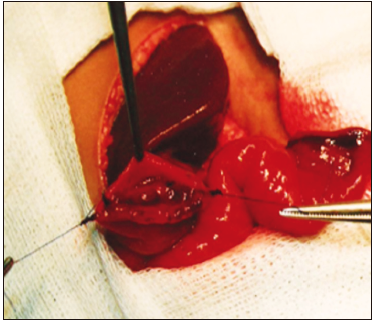
Figura 3. Incisión subcostal derecha y ostomía en fosa ilíaca derecha finalizada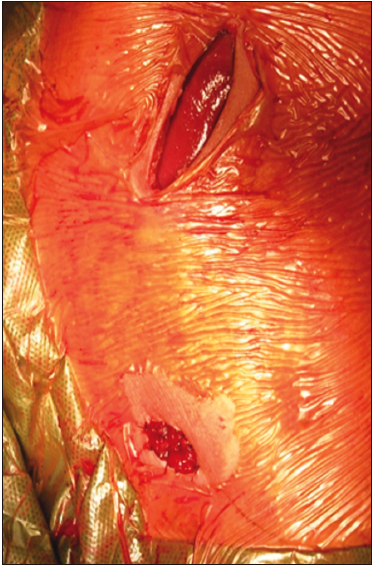
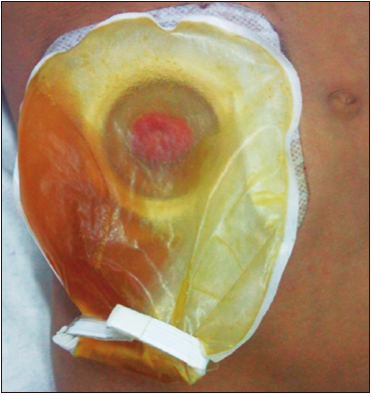
Figura 4. Ostomía funcionante drenando solo bilis
El diagnóstico de PFIC se realizó en un paciente por mutación genética compatible con PFIC2 y en los cuatro restantes por las características clínicas, bioquímicas e histológicas, descartándose otras causas de colestasis. En los pacientes con SA se estableció por la presencia de tres o más criterios mayores.
El rango de edad al momento del diagnóstico fue de 45 días de vida a 6 meses, con una mediana de 2,5 meses (Tabla 2). Siete de los ocho pacientes presentaron colestasis neonatal como manifestación inicial de la patología de base. Todos los pacientes presentaban prurito refractario al tratamiento médico y mal progreso pondoestatural antes del procedimiento.
Tabla 2. Características de los pacientes 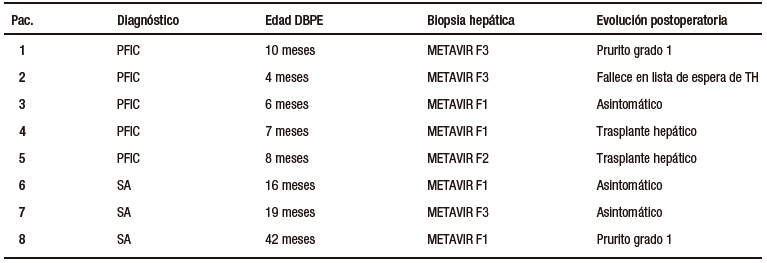
En los pacientes con PFIC las biopsias hepáticas previas a la cirugía informaron fibrosis severa en dos casos, fibrosis moderada en uno y fibrosis leve en dos. Los pacientes con SA presentaron fibrosis severa en un caso, y leve en los dos restantes.
Todos los pacientes con PFIC fueron sometidos a la DBPE antes del año de vida. En los pacientes con SA la DBPE fue realizada a los 16, 19 y 42 meses de vida, luego de haber fracasado el tratamiento médico para el manejo del prurito.
Ningún paciente presentó complicaciones inmediatas secundarias al procedimiento. Un paciente con diagnóstico de PFIC presentó sangrado por el ostoma, diecinueve años después de la cirugía, cuya causa fue una mucositis erosiva. Una paciente con SA fue reintervenida a los cuatro meses para rehacer la ostomía, debido a que había quedado estrecha.
Dos de los cinco pacientes con PFIC presentaron evolución favorable con mejoría del patrón colestásico en el laboratorio al mes del procedimiento y laboratorios estables hasta la actualidad. Uno de ellos resolvió completamente el prurito a los tres meses de la DBPE y el otro presentó resolución parcial, desde los 14 días posquirúrgicos, pasando de grado 3 a grado 1. Los otros tres pacientes presentaron progresión de la enfermedad, por lo cual se indicó el trasplante hepático a los 13 y 15 meses de realizada la DBPE, respectivamente.
Dos de los pacientes con SA presentaron una buena evolución clínica y bioquímica. El perfil lipídico y el patrón colestásico mejoraron progresivamente entre los tres y cinco meses posteriores al procedimiento. En un solo caso persiste el prurito grado 1, y el único paciente con xantomas previos a la cirugía presentó involución gradual hasta su completa resolución. Ningún paciente con SA requirió trasplante hepático (Tabla 3).
Tabla 3. Evolución clínica y bioquímica. Prurito según escala de Whittington 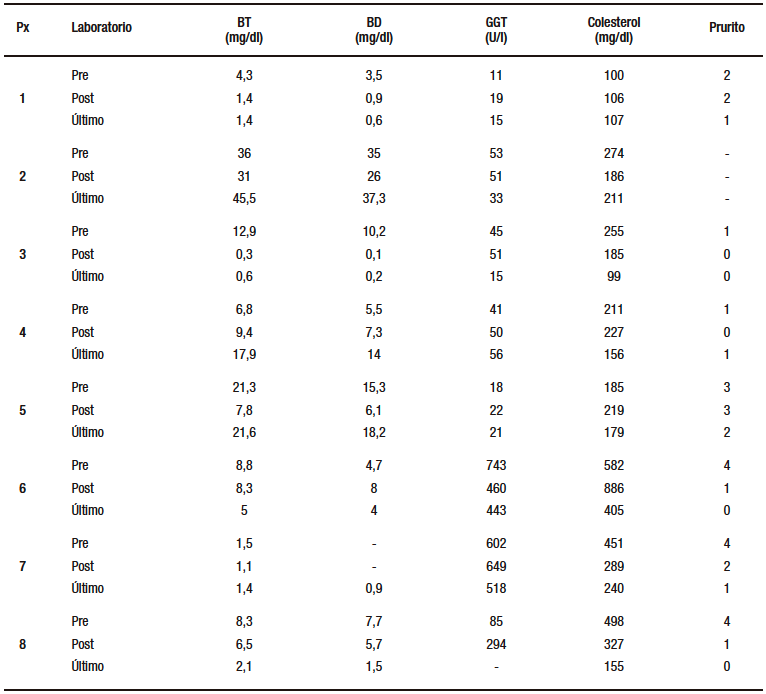
Discusión
La DBPE, descrita por Whittington en el año 1988, es una de las técnicas quirúrgicas que tiene como objetivo realizar una derivación del flujo biliar, con la intención de disminuir la bilis ofertada para la recirculación enterohepática de las sales biliares, evitando así su acumulación en el hepatocito y el consiguiente daño hepático.5 Se utilizó inicialmente en pacientes con PFIC 1 y 2, pero su uso se extendió a pacientes con colestasis crónica por SA, con resultados favorables. Esta técnica debe realizarse previo al desarrollo de cirrosis, dado que en pacientes cirróticos no ha mostrado resultados favorables.6, 7 En los pacientes con PFIC 1 y 2, la DBPE constituye un tratamiento alternativo, ya que demostró mejorar el prurito y los niveles plasmáticos de ácidos biliares, enlentecer el progreso de la enfermedad hepática y mejorar la histopatología en el 85% de los pacientes.5 Se postula que el principal efecto de la DBPE en estos pacientes es la modificación del pool de ácidos biliares por una composición mayormente hidrofílica, que genera menor daño celular y la mejoría de la colestasis.6, 8
En el caso de nuestros pacientes con PFIC, observamos una evolución variable que no parece correlacionarse con el compromiso histológico previo a la cirugía. De los dos pacientes con fibrosis severa, uno requirió trasplante por progresión de la enfermedad y el otro presentó buena evolución clínica y luego de 21 años de seguimiento fue derivado a un equipo de adultos. La gran diversidad genética y la presencia de mutaciones homocigotas en los genes involucrados podría ser un factor de peor pronóstico en comparación con pacientes con heterocigosis en estos u otros genes. El paciente con PFIC tipo 2 que tuvo resolución del prurito y normalización del laboratorio presenta una mutación heterocigota del gen ABCB11.
En todos nuestros pacientes con SA, la DBPE demostró mejorar el prurito, el perfil lipídico y los xantomas, en concordancia con lo descripto en la literatura.9 La mejoría en la calidad de vida cambia favorablemente el pronóstico de los pacientes, evitando o retrasando la indicación de trasplante hepático.
Es importante destacar que la DBPE no invalida la realización de un trasplante hepático en el caso de ser necesario a posteriori. Si bien se han descrito complicaciones de la cirugía, como el prolapso, el sepultamiento del ostoma10 o el excesivo drenaje de bilis, con la consiguiente malabsorción y alteración electrolítica,9 esta cirugía se considera un procedimiento seguro, sin mortalidad reportada,10 cuyos beneficios permiten definirla como una opción terapéutica. En nuestra serie solo dos pacientes presentaron complicaciones, tal como se mencionó en los resultados.
La fístula biliocutánea (ostomía) suele ser un motivo de rechazo de la cirugía; sin embargo, en nuestra experiencia, observamos que tanto los pacientes como sus familias lograron adaptarse a ella y a sus cuidados, pudiendo mantener su calidad de vida, la escolarización y la socialización. En este sentido, se han desarrollado procedimientos de derivación biliar interna (DBI) en el intento de limitar la recirculación de sales biliares sin la utilización de una fístula cutánea, drenando la bilis al colon, pero, hasta la fecha, no se cuenta con comparaciones concluyentes entre los resultados a largo plazo de ambas técnicas;9, 11 se reporta diarrea osmótica o malabsortiva como complicación frecuente de la DBI, y se desconoce el efecto a largo plazo de la exposición colónica a la bilis, e incluso se observó la recurrencia de la sintomatología en algunas series.11, 12 También se describe la utilización del apéndice como conducto de derivación, aunque no ha alcanzado aceptación, al igual que los distintos tipos de by pass de la región ileocecal.
Actualmente, se encuentran en desarrollo estudios con inhibidores selectivos y reversibles de transportadores de ácidos biliares ileales administrados por vía oral como alternativa de tratamiento farmacológico en pacientes con PFIC 1 y 2, y como tratamiento para el control del prurito en SA de forma no invasiva.13 Los resultados de estos estudios a largo plazo determinarán su seguridad y eficacia con respecto a la mejoría del prurito, la colestasis y la evolución histológica, para poder ser considerados como tratamientos de elección.
Conclusiones
La eficacia de la DBPE en mejorar la calidad de vida y, en algunos casos, enlentecer o detener la progresión de la enfermedad hepática está claramente demostrada en pacientes con PFIC 1 y 2 y SA, y debe ser considerada en ausencia de otro tratamiento alternativo.
La cirugía tiene una baja morbilidad y no impide la realización ulterior de un trasplante hepático, si se lo requiriera.
La DBPE mejoró la calidad de vida y las complicaciones asociadas a la colestasis crónica en la mayoría de nuestros pacientes, evitando el trasplante hepático en cinco de ellos (dos PFIC y tres SA) y difiriéndolo en dos.
Esta alternativa quirúrgica debe ser tenida en cuenta en el manejo terapéutico precoz de pacientes con colestasis genéticas y requiere un manejo multidisciplinario en un centro pediátrico con experiencia en enfermedades hepáticas y cirugía hepatobiliar.
Consentimiento para la publicación. Para la confección de este manuscrito, se utilizaron datos anonimizados que no han distorsionado su significado científico.
Propiedad intelectual. Los autores declaran que los datos, las tablas y las figuras presentes en el manuscrito son originales y fueron realizados en sus instituciones pertenecientes.
Financiamiento. Los autores declaran que no hubo fuentes de financiación externas.
Conflicto de interés. Los autores declaran no tener conflictos de interés en relación con este artículo.
Aviso de derechos de autor

© 2022 Acta Gastroenterológica Latinoamericana. Este es un artículo de acceso abierto publicado bajo los términos de la Licencia Creative Commons Attribution (CC BY-NC-SA 4.0), la cual permite el uso, la distribución y la reproducción de forma no comercial, siempre que se cite al autor y la fuente original.
Cite este artículo como: Salgueiro F, Lezama Elecharri C, Galoppo M y col. Derivación biliar parcial externa en colestasis intrahepática familiar progresiva y síndrome de Alagille. Serie de casos. Acta Gastroenterol Latinoam. 2022;52(3):388-394. https://doi.org/10.52787/agl.v52i3.230
Referencias
- Balistreri WF, Bezerra JA. Whatever Happened to “Neonatal Hepatitis”? Clinics in Liver Disease. 2006;27-53.
- Devit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet Journal of Rare Diseases. 2009;4(1).
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clinics and Research in Hepatology and Gastroenterology. 2012;36:S26-S35.
- Emerick KM, Whitington PF. Partial External Biliary Diversion for Intractable Pruritus and Xanthomas in Alagille Syndrome. Hepatology. 2002;35(6).
- van del Woerd WL, Houwen RH, van der Graaf SF. Current and future therapies for inherited cholestatic liver diseases. World Journal of Gastroenterology. 2017;23(5):763-75.
- Smith Jericho H, Kaurs E, Boverhof R, Knisley A, Shneider BL, Verkade HJ, et al. Bile Acid Pool Dynamics in Progressive Familial Intrahepatic Cholestasis with Partial External Bile Diversion. J Pediatr Gastroenterol Nutr. 2015;60(3):368-74.
- Srivastava A. Progressive Familial Intrahepatic Cholestasis. Journal of Clinical and Experimental Hepatology. 2014;4(1):25-36.
- Emerick KM, Elias MS, Melin-Aldana H, Strautnieks S, Thompson RJ, Bull LN, et al. Bile composition in Alagille Syndrome and PFIC patients having Partial External Biliary Diversion. BMC Gastroenterology. 2008;8(47).
- Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, et al. Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017;65(5):1645-54.
- Yang H, Porte RJ, Verkade HJ, De Langen Z, Hulscher JBF. Partial External Biliary Diversion in Children With Progressive Familial Intrahepatic Cholestasis and Alagille Disease. Journal of Pediatric Gastroenterology and Nutrition. 2009;49(2):216-21.
- Jankowska I, Czubkowski P, Kalicinski P, Ismail H, Kowalski A, Ryzko J, et al. Ileal Exclusion in Children With Progressive Familial Intrahepatic Cholestasis. Journal of Pediatric Gastroenterology and Nutrition. 2014;58:92-5.
- Kalicinski PJ, Ismail H, Jankowska I, Kaminski A, Pawlowska J, Drewniak T, et al. Surgical Treatment of Progressive Familial Intrahepatic Cholestasis: Comparison of Partial External Biliary Diversion and Ileal Bypass. Eur J Pediatr Surg. 2003;13:307-11.
- Slavetinsky C, Strum E. Odevixibat and partial external biliary diversion showed equal improvement of cholestasis in a patient with progressive familial intrahepatic cholestasis. BMJ Case Rep. 2020;13:e234185.
Correspondencia: Carol Lezama Elecharri
Correo electrónico: lezamacarol@hotmail.com
Acta Gastroenterol Latinoam 2022;52(3):388-394
