Emanuel José Saad,1, 2 María Victoria Mulqui,1 Candelaria Mendoza,3 Juliana Mandrile,4 Domingo Cesar Balderramo2, 5
1Internal medicine department, Hospital Privado Universitario de Córdoba.
2Instituto Universitario de Ciencias Biomédicas de Córdoba.
3Pathology Department, Hospital Privado Universitario de Córdoba.
4Hematology and Oncology Department, Hospital Privado Universitario de Córdoba.
5Gastroenterology Department, Hospital Privado Universitario de Córdoba.
Córdoba, Argentina.
Acta Gastroenterol Latinoam 2018;48(4):322-326
Recibido: 17/11/2017 / Aprobado: 14/12/2017 / Publicado en www.actagastro.org el 17/12/2018

Summary
POEMS syndrome is a rare paraneoplastic disorder with an underlying plasma cell dyscrasia associated with peripheral polyneuropathy, organomegaly, endocrinopathy, monoclonal gammapathy and skin changes. While, ascites is a common sign in patients with POEMS, it is unfrequently associated with portal cavernomatosis and portal hypertension. We report a case of a 41-year-old man who presented with asthenia, weight loss and ascites. There was diagnosed portal cavernomatosis and the ascitic fluid exam was compatible with transudate. Later, the patient developed polyneuropathy and cutaneous hyperpigmentation, and there were demonstrated monoclonal component IgA in serum, Castleman disease in an inguinal lymphadenophaty and osteosclerotic injuries in bones, and the patient was diagnosed with POEMS syndrome. Only 5 cases of POEMS syndrome with portal hypertension have been reported. Nevertheles, in anyone of them portal thrombosis was demonstrated, and there was described only one case of a patient with POEMS syndrome, who developed portal thrombosis.
Key words. POEMS syndrome, polyneuropathies, paraproteinemias, venous thrombosis.
Reporte de caso. Cavernomatosis portal como presentación de un síndrome de POEMS
Resumen
El síndrome de POEMS es una infrecuente manifestación paraneoplásica de las discrasias de células plasmáticas que se encuentra asociada a polineuropatía periférica, organomegalia, endocrinopatía, gammapatía monoclonal y alteraciones en la piel. Si bien la ascitis es un signo común en los pacientes con síndrome de POEMS, es muy infrecuente su asociación con cavernomatosis e hipertensión portal. Presentamos el caso de un hombre de 41 años que se presentó con astenia, pérdida de peso corporal y ascitis. Se realizó diagnóstico de cavernomatosis portal y el examen de líquido ascítico fue compatible con trasudado. Posteriormente el paciente desarrolló polineuropatía e hiperpigmentación cutánea. Se demostró la presencia de gammapatía monoclonal IgA en suero, enfermedad de Castleman en una adenopatía inguinal, además de lesiones osteoescleróticas en los huesos, por lo que se realizó el diagnóstico de síndrome de POEMS. Hasta la fecha solamente se han reportado 5 casos de síndrome de POEMS asociado a hipertensión portal. Sin embargo, en ninguno de ellos se han observado signos compatibles con cavernomatosis portal, habiéndose descripto solo un caso de trombosis portal en pacientes con síndrome de POEMS.
Palabras claves. Síndrome POEMS, polineuropatías, paraproteinemias, trombosis de la vena.
Abbreviations
NV: normal values.
SAAG: serum-ascites albumin gradient.
CRP: c-reactive protein.
VEGF: vascular endothelial growth factor.
POEMS syndrome is an infrequent paraneoplastic disorder of plasma cell dyscrasia. The acronym ‘POEMS’ was given to the disorder on the basis of its main clinical characteristics: Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal component, and Skin changes.1 Their low incidence and slow progression is associated to a diagnosis delay of approximately 13 to 18 months.2 While ascites is a common sign in patients with POEMS, the presence of portal cavernomatosis and portal hypertension is extremely infrequent.
Case report
We present a case of a 41-year-old male with weakness in lower limbs, asthenia, and a 10% body weight loss during the last year, associated with night sweating. He had not experienced fever neither any other related symptoms. The patient had history of lung tuberculosis at age 24, which had been successfully treated. There was no history of drugs, tobacco or alcohol abuse.
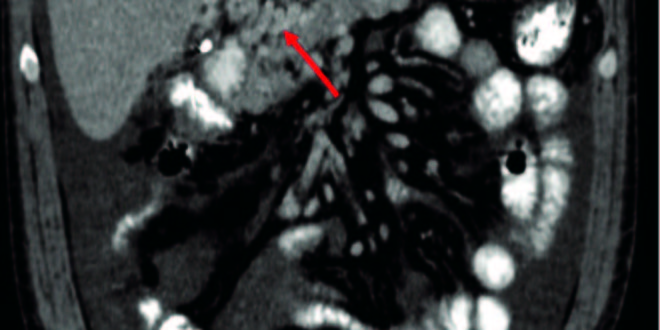
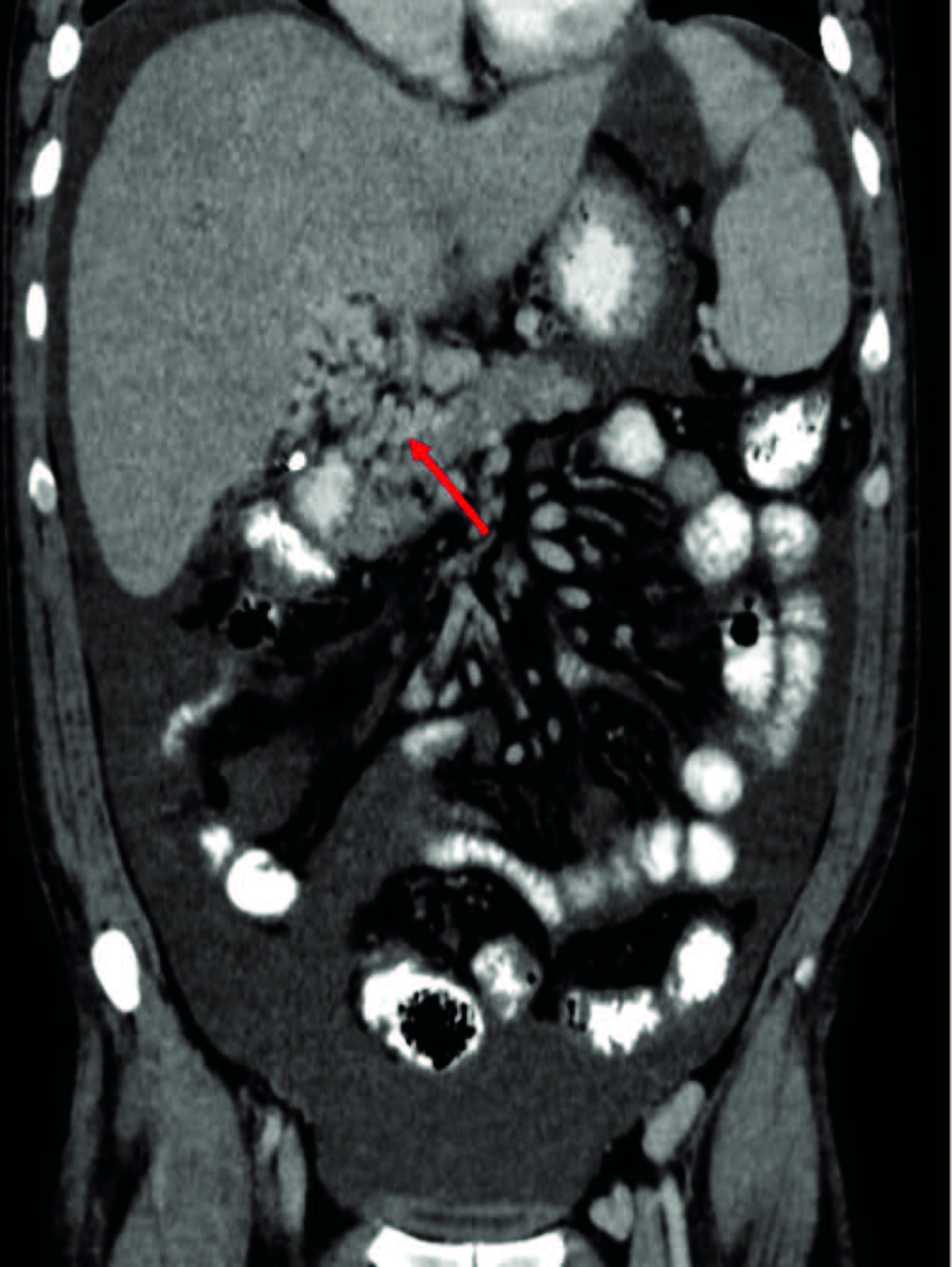
On physical examination, moderate abdominal ascites and hepatosplenomegaly was present only. A computed tomography of the thorax and abdomen showed the presence of hepatosplenomegaly, portal cavernoma with abundant ascitic fluid and a 15mm adenomegaly located in the right inguinal region (Figure 1). Upper endoscopy showed grade III esophageal varices and portal hypertensive gastropathy. Full blood count, renal function and hemostasis test were normal. Liver test exam showed: bilirubin of 1.39 mg/dL, aspartate-aminotransferase 59 U/L (NV < 37U/L), alanine-aminotransferase 60 U/L (NV < 41U/L), gamma-glutamyl-transpeptidase 377 U/L(NV < 49 U/L), alkaline phosphatase 756 U/L (NV: 91-258 U/L), lactate dehydrogenase of 184 U/L(NV: 236-460U/L) and erythrocyte sedimentation rate of 43 mm/Hr. Iron metabolism, ceruloplasmin and alpha-1 antitrypsin were also normal. Serologic tests for human immunodeficiency virus, hepatitis B virus, hepatitis C virus, VDRL, Chagas disease, anti-smooth muscle, antimitochondrial, anticytoplasmic and anti-nuclear antibodies were negative. Ascitic fluid exam showed a serum-ascites albumin gradient (SAAG) compatible with transudate fluid (16g/L), cell count of 710/mm3 with mononuclear predominance, and culture were negatives. An hepatic biopsy yielded mild inflammatory infiltrate on the portal region without fibrosis. Thrombophilia screening showed normal values of C and S proteins, absence of lupus anticoagulants, JAK2 mutation, prothrombin gene mutation 20210, Leiden V factor, IgG and IgM anti-cardiolipin, anti-beta2 microglobulin and anti-thrombin III antibodies. Treatment with diuretics was started.
Figura 1. Abdominal computed tomography scan. Coronal section showed hepatoesplenomegaly and a portal cavernoma and multiple tortuous vessels and ascites.
After the initial evaluation, the patient experienced progressive deterioration over the following 4 months. Paresthesia and progressive weakness were presented in his lower limbs. There was progression of ascites, lower limbs edemas and developed skin hyperpigmentation, with predominance on the face, extremities, and mucous membranes. Physical examination showed weakness, in both, tibialis anterior and extensor hallucis muscles, steppage gait, areflexia in Achilles´s tendons and distal hypopallesthesia, withouth upper limbs involvement. A lower limbs electromyography was performed showing a sensitive-motor axonal polyneuropathy. B12 vitamin and folate level was 322pg/ml and 9.0ng/ml respectively, while plasmatic cortisol was 7.9 μg/dl and adrenocorticotropin was 113 pg/ml.
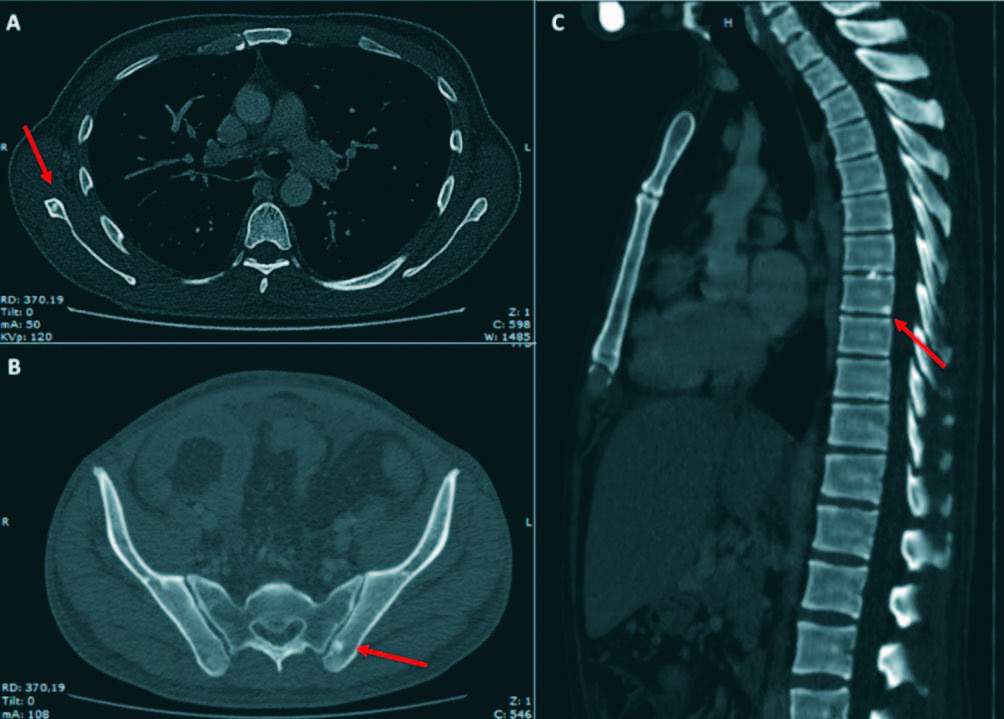
Due to the absence of etiology diagnosis, a laparotomy was conducted, hepatic and peritoneal biopsies were performed and the left inguinal lymphadenophaty was excised. Their mycrobiological cultures and CRP for Mycobacterium tuberculosis and Histoplasma capsulatum were negative. The patho-logy analysis of the inguinal lymphadenophaty revealed the presence of vessels in central position, surrounded by lymphocytes in crown-like structures, reminiscent of the hyaline vascular Castleman disease (Figure 2). The hepatic and peritoneal biopsy showed normal findings and amyloid deposit was negative in both biopsies. The serum immunofixation test showed an IgA lambda biclonal band, while urine serum test was negative. The free kappa and lambda light chains concentrations were 67.31 mg/l (NV: 3.30-19.40) and 84.36 (NV: 5.71-26.30) respectively, with a kappa/lambda ratio of 0.8 (NV: 0.26-1.65). A bone marrow biopsy showed hyperplasia and dysplastic changes, with erythroid predominance and mild plasmacytosis of 4.2%, of which 70.3% were aberrant (CD138+, CD38+, CD19-, CD45- , CD56+), compatible with plasma cells dyscrasia. After reviewing the previous computed tomography images, several osteosclerotic lesions were observed on the vertebral column, the scapula, and the hip bone. (Figure 3) Owing to the identification of polyneuropathy (compatible with axonal compromise), organomegaly (hepatosplenomegaly), potential endocrinopathy (indicative of suprarenal insufficiency), monoclonal component IgA Lambda, cutaneous hyperpigmentation, Castleman disease and osteosclerotic injuries, the patient was diagnosed with POEMS syndrome after 20 months of diagnostic evaluation.
The patient underwent glucocorticoids substitution therapy and chemotherapy with Cyclophosphamide dexamethasone for 6 cycles, showing good clinical re sponse, progressive muscular strength recovery and reduction of the edematous syndrome.
Figura 2. Lymph node biopsy compatible with Castleman disease, hyaline vascular type. A) Showed regressed follicles with expanded mantle zone showing a “onion skin” pattern (Haematoxylin and eosin stain, original magnification x4). B) Depleted and hyalinized germinal center with increased vascularity penetrating radiated (Haematoxylin and eosin stain, original magnification x40).
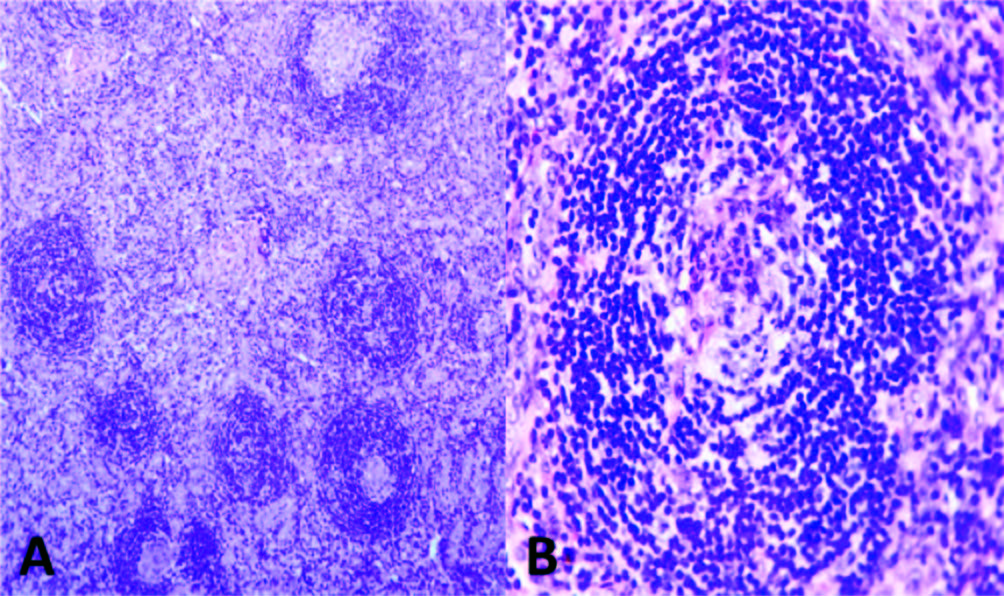
Figura 3. Thoracic and abdominal computed tomography scan. Osteosclerotic bone lesions are noticed in the right scapular (A), left iliac bone (B) and the inferior angle of vertebral body D8 (C). Coronal section showed hepatoesplenomegaly wuth and a portal cavernoma with multiple tortuous vessels and ascites (D).
Discussion
The diagnosis of POEMS syndrome is confirmed when the patient meets diagnostic criteria, which are classified into major and minor. Within the major criteria, polyneuropathy and the monoclonal component are mandatory to diagnose the syndrome. Other major criteria includes: Castleman disease, osteosclerotic inju ries and the elevated vascular endothelial growth factor (VEGF). Minor criteria includes organomegaly (splenomegaly, hepatomegaly or lymphadenopathy), extravascular volume overload, endocrinopathy, skin injuries (such as hyperpigmentation and leuconychia), papilledema and thrombocytosis and/or polycythemia. In order to be able to confirm diagnosis, the presence of the both mandatory criteria plus at least one of the remaining major criteria and one of the minor criteria is required.3 The pathogenesis of POEMS syndrome has not yet been completely clarified, but it is believed that many factors are involved including a high production of interleukin-1beta, Interleukin-6, TNF-alpha, and the vascular endothelial growth factor (VEGF).4
Extravascular volume overload is one of the most common manifestations of POEMS, characterized by peripheral edemas, pleural effusion, ascites, and pericardial effusion. The association between ascites and POEMS is present in 23-48% of patients; however, the available information on the pathogenesis and the physico-chemical characteristics of ascitic fluid in those patients remains scarce. One of the largest series of studies on ascitic fluid in POEMS patients was conducted by Cui et al.5 In this study that include 106 patients with POEMS, 39% of them presented ascites. All patients had SAAG < 11g/L, which is compatible with exudate, suggesting an abnormal permeability of capillaries in the visceral peritoneum. In addition, there were no cases portal thrombosis or hepatic vein thrombosis according to Doppler evaluation.5 Only 5 cases of POEMS syndrome with portal hypertension have been reported, four of them with evidence of non-cirrhotic portal fibrosis,6-10 and one with unknown mechanism.11 Nevertheles, in none of them portal thrombosis was demonstrated. Although POEMS patients have an increased risk of venous and arterial thrombosis of about 20%, there are no cases that reported portal cavernomatosis as a manifestation.2, 6, 12 There was described only one case of a patient with POEMS syndrome, who developed portal thrombosis during the chemotherapy treatment,13 and there are few cases associated to Budd- Chiari syndrome.14, 15 Several factors as progressive fibrosis of the portal veins, acquired vascular defect, exposure to several toxins or infections, immunological basis, aberrations in the thrombin-antithrombin complex and elevations of inflammatory cytokines, such as VEGF, Interleukin-1β, Interleukin-6, Interleukin-12 and tumor necrosis factor-α _that are increased in POEMS syndrome may be related with the underlying mechanism of POEMS syndrome and portal hypertension.11
Another important fact in this case is the coexistence with Castleman disease, a condition presented in 11%- 30% of patients with POEMS syndrome.16 Clinical manifestations vary greatly according to its histological type and extension, being a challenge performing the diagnosis. It is frequently associated with anemia and/or thrombocytopenia, unlike POEMS, which usually pre-sent thrombocytosis and polycythemia. The abdominal-pelvic region is affected in only 15% of all patients.17
The average survival of POEMS syndrome is about 14 years. The association with Castleman disease is generally related to a worse prognosis. The treatment of the disease depends on the extension of plasma cells infiltration. It is considered a spread disease if it involves bone marrow and/or more than three skeletal lesions. High doses of chemotherapy with autologous transplant of stem cells have also proved to be effective (100% of all surviving patients have experienced improved symptoms).3
In conclusion, POEMS syndrome is an extremely rare disease and may include a wide variety of clinical manifestations. The presentation as ascitic syndrome with portal hypertension is very rare, and no cases of portal hypertension associated to portal cavernomatosis, as the present case, have been reported.
Conflicts of interest. The authors declare that there are no conflicts of interest and there are no founding source.
Acknowledgements. Marcela Colazo, Ruth Kaplan.
References
- Andresik D, Russo MP. Síndrome POEMS. Comunicación de cuatro casos. Medicina (B Aires) 2015; 75: 324-327.
- Dispenzieri A, Buadi FK. A review of POEMS syndrome. Oncology (Williston Park) 2013; 27: 1242-1250.
- Dispenzieri A. POEMS syndrome: update on diagnosis, risk-stratification, and management. Am J Hematol 2015; 90: 951-962.
- Marinho FS, Pirmez R, Nogueira R, Cuzzi T, Sodré CT, Ramos-e-Silva M. Cutaneous Manifestations in POEMS Syndrome: Case Report and Review. Case Rep Dermatol. 2015; 7: 61-69.
- Cui RT, Yu SY, Huang XS, Zhang JT, Li F, Pu CQ. The characteristics of ascites in patients with POEMS syndrome. Ann Hematol 2013; 92: 1661-1664.
- Varona Arche JF, Aranda Arcas JL. Hipertensión portal por cavernomatosis esplenoportal. An Med Interna 2005; 22: 93-94.
- Inoue R, Nakazawa A, Tsukada N, Katoh Y, Nagao T, Nakanuma Y, Mukai K. POEMS syndrome with idiopathic portal hypertension: autopsy case and review of the literature. Pathol Int 2010; 60: 316-320.
- Teramoto J, Itoh R, Inagaki T, Mukoyama M, Takeuchi J. A case of polyneuropathy with monoclonal gammopathy, hepatosplenomegaly, ascites, skin pigmentation, and swelling of salivary glands. Neurol Med 1977; 7: 500-503.
- Stepani P, Courouble Y, Postel P, Mezieres P, Tossou H, Couvelard A, Trophilme D, Barbare JC, Bories C. Portal hypertension and neutrocytic ascites in POEMS syndrome. GastroenterolClin Biol 1998; 22: 1095-1097.
- Campos S, Agostinho C, Cipriano MA. POEMS syndrome and idiopathic portal hypertension: a possible association. Rev Esp Enferm Dig 2017; 109: 393.
- Wu L, Li Y, Yao F, Lu C, Li J, Zhou W, Qian J. Portal hypertension as the initial manifestation of POEMS syndrome: a case report. BMC Hematol 2017; 17: 9.
- Kumar A, Sharma P, Arora A. Review article: portal vein obstruction–epidemiology, pathogenesis, natural history, prognosis and treatment. Aliment Pharmacol Ther 2015; 41: 276-292.
- Ganti AK, Pipinos I, Culcea E, Armitage JO, Tarantolo S. Successful hematopoietic stem-cell transplantation in multicentric Castleman disease complicated by POEMS syndrome. Am J Hematol 2005; 79: 206-210.
- Dispenzieri A, Kyle RA, Lacy MQ, Rajkumar SV, Therneau TM, Larson DR, Greipp PR, Witzig TE, Basu R, Suárez GA, Fonseca R, Lust JA, Gertz MA. POEMS syndrome: Definitions and long-term outcome. Blood 2003; 101: 2496-2506.
- Almenta IM, Martínez B, Palazón JM. Budd-Chiari syndrome as clinical expression of POEMS syndrome. Gastroenterol Hepatol 2015; 38: 380-382.
- Andhavarapu S, Jiang L. POEMS syndrome and Castleman disease. Blood 2013; 122: 159.
- Cervantes CE, Correa R. Castleman Disease: A Rare Condition with Endocrine Manifestations. Cureus 2015; 7: e380.
Correspondencia: Emanuel José Saad
Internal Medicine Department, Hospital Privado Universitario de Córdoba, Instituto Universitario de Ciencias Biomédicas de Córdoba. Av. Naciones Unidas 346. (CPA X5016KEH). Córdoba, Argentina
Tel.: (+54) 351 4688245 / Fax: (+54) 351 4688875
Correo electrónico: emanuelsaad@hotmail.com
Acta Gastroenterol Latinoam 2018;48(4): 322-326
