María Laura Moreno
Médica gastroenteróloga. Sección Intestino Delgado. Hospital de Gastroenterología “Dr. Carlos Bonorino Udaondo”.
Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2020;50(4):388-414
Recibido: 09/11/2020 / Aprobado: 27/11/2020 / Publicado en www.actagastro.org el 14/12/2020

Resumen
La diarrea crónica constituye un motivo frecuente de la derivación al especialista y su prevalencia se estima en alrededor del 4 al 5%. Llegar a un diagnóstico etiológico es un verdadero desafío clínico. Los mecanismos fisiopatológicos involucrados en su génesis son múltiples y, a veces, coexisten en un mismo individuo. Una anamnesis correctamente orientada y el examen físico permitirán identificar datos claves, principalmente la presencia de síntomas o signos de alarma con la intención de discriminar entre las causas funcionales y las orgánicas. El médico debe saber utilizar su criterio clínico, evaluar las probabilidades y utilizar los métodos diagnósticos disponibles con la orientación precisa para adecuarlos a cada caso en particular. La categorización de la diarrea en acuosa, malabsortiva o inflamatoria intenta simplificar el proceso diagnóstico, si bien es solo orientativa. Es importante conocer las distintas patologías que se presentan con diarrea crónica, cuándo sospecharlas y cómo llevar adelante el proceso diagnóstico de manera costo-efectiva así como también los posibles desencadenantes (medicamentos, alimentos, trastornos endocrinológicos, tumores, radiación, etc.). El objetivo de la siguiente revisión es dar a conocer los diferentes mecanismos fisiopatológicos involucrados en la diarrea crónica, así como las herramientas diagnósticas y terapéuticas disponibles que nos permitan realizar un enfoque adecuado para su estudio.
Palabras claves. Diarrea crónica, diarrea funcional, síndrome del intestino irritable, malabsorción, esteatorrea, diarrea secretora, enfermedad celíaca, enfermedad inflamatoria intestinal, pruebas diagnósticas.
Diagnostic Approach to Chronic Diarrhea
Summary
Chronic diarrhea is a frequent reason for the referral to the specialist and its prevalence is estimated at around from 4 to 5%. Reaching an etiological diagnosis is a real clinical challenge. The pathophysiological mechanisms involved in its genesis are multiple and sometimes coexist in the same individual. A correctly oriented anamnesis and physical examination will allow to identify key data, mainly the presence of symptoms or alarm signs with the intention of discriminating between the functional and the organic causes. The doctor must know how to use his clinical judgment, evaluate the probabilities and use the diagnostic methods available with the precise orientation to adapt them to each particular case. The categorization of diarrhea as watery, malabsorptive or inflammatory attempts to simplify the diagnostic process, although it is only indicative. The objective of the following review is to present the different pathophysiological mechanisms involved in chronic diarrhea, as well as the available diagnostic and therapeutic tools that allow us to carry out an adequate approach for its study.
Key words. Chronic diarrhea; functional diarrhea; irritable bowel syndrome; malabsorption, steatorrhea, secretory diarrhea, coeliac disease, inflammatory bowel disease, diagnostic testing.
Abreviaturas
HC: Historia clínica.
CCR: Cáncer colorrectal.
EII: Enfermedades inflamatorias intestinales.
EC: Enfermedad celíaca.
AINEs: Antiinflamatorios no esteroideos.
ATB: Antibióticos.
DBT: Diabetes.
FODMAPS: Oligosacáridos, disacáridos, monosacáridos y polioles fermentables.
STROKE: Accidente cerebro vascular.
VIC: Válvula ileocecal.
SB: Sales biliares.
SBID: Sobrecrecimiento bacteriano del intestino delgado.
ESD: Eritrosedimentación.
PCR: Proteína C reactiva.
AG: Anión gap.
CPF: Calprotectina fecal.
AGCC: Ácidos grasos de cadena corta.
SGNC: Sensibilidad al gluten no celíaca.
SII-D: Síndrome del intestino irritable variedad diarrea.
DF: Diarrea funcional.
TSOMF: Test de sangre oculta en materia fecal.
IBP: Inhibidores de la bomba de protones.
IRC: Insuficiencia renal crónica.
EIA: Enzimoinmunoanálisis.
GDH: Glutamato deshidrogenasa.
EA: Enteritis actínica.
MAB: Malabsorción de los ácidos biliares.
DCL: Deficiencia congénita de lactasa.
IL: Intolerancia a la lactosa.
TAE: Test del aire espirado.
ID: Intestino delgado.
JMAF: Jarabe de maíz de alta fructosa.
CM: Colitis microscópica.
CC: Colitis colágena.
CL: Colitis linfocítica.
TNE: Tumores neuroendocrinos.
IRC: Insuficiencia renal crónica.
AR: Artritis reumatoidea.
VCC: Videocolonoscopía.
RSC: Rectosigmoideoscopía.
LIEs: Linfocitos intraepiteliales.
RM: Resonancia magnética.
TC: Tomografía computada.
TNE-GEP: Tumores neuroendocrinos gastroenteropancreáticos.
PET/TC: Tomografía por emisión de positrones.
CgA: Cromogranina A.
VIP: Péptido intestinal vasoactivo.
SST: Somatostatina.
SZE: Síndrome de Zollinger Ellison.
G: Gastrinemia.
5-HIAA: Ácido 5 hidroxindolacético.
SS: Somatostatina.
MTS: Metástasis.
T: Tumor.
HTA: Hipertensión arterial.
NEM: Neoplasia endócrina múltiple.
IPE: Insuficiencia pancreática exocrina.
H: Hidrógeno.
CH4: Metano.
CA: Ácido cólico.
CDCA: Ácido quenodesoxicólico.
ADC: Ácido desoxicólico.
5-HT: Serotonina.
ALC: Ácido litocólico.
TBC: Tuberculosis.
RT: Radioterapia.
PC: Pancreatitis crónica.
FXR: Receptor farsenoide X.
IDCV: Inmunodeficiencia común variable.
EW: Enfermedad de Whipple.
VIH: Virus de inmunodeficiencia humana.
EAI: Enteropatía autoinmune.
T1DM: Diabetes mellitus tipo 1.
TG2: Transglutaminasa 2.
E: Especificidad.
S: Sensibilidad.
NBI: Narrow-band imaging.
FICE: Fujinon intelligent chromo endoscopy system.
AV: Atrofia vellositaria.
EC: Enfermedad de Crohn.
CU: Colitis ulcerosa.
LES: Lupus eritematoso sistémico.
PTI: Púrpura trombótica idiopática.
LNH: Linfoma no Hodgkin.
GVHD: Enfermedad de injerto contra huésped aguda.
GE Eo: Gastroenteritis eosinofílica.
HPF: Campos de gran aumento.
IFN: Interferón gamma.
SNC: Sistema nervioso central.
ANCA: Anticuerpos anticitoplasma de neutrófilos.
ELISA: Enzimoinmunoanálisis de adsorción.
SC: Esprúe colágeno.
CMV: Citomegalovirus.
CD: Clostridium difficile.
ICD: Infección por Clostridium difficile.
ICC: Células intersticiales de Cajal.
SNE: Sistema nervioso entérico.
NANC: No adrenérgica no colinérgica.
ON: Óxido nítrico.
CL a1 AT: Clearence de alfa 1 antitripsina.
VEDA: Videoendoscopía digestiva alta.
VCE: Videocápsula endoscópica.
MF: Materia fecal.
TSH: Tirotrofina.
VPN: Valor predictivo negativo.
CTNA: Ensayo de neutralización de citotoxina.
NAAT: Técnicas de amplificación de ácidos nucleicos.
IGIV: Inmunoglobulina endovenosa.
UCI: Unidad de cuidados intensivos.
SIRS: Inhibidores de la recaptación de serotonina.
ADEK: Vitaminas liposolubles A, D, E y K.
ACG: Sigla American College of Gastroenterology.
CT: Cultivo toxigenico.
CCNA: Cell Cytotoxicity Neutralization Assay Prueba de neutralización de citotoxicidad.
Introducción
Se define a la diarrea crónica como a la alteración persistente de la forma de las heces, consideradas las consistencias entre los tipos 5 a 7 de la Escala de Bristol y al aumento en la frecuencia de las deposiciones de una duración mayor a 4 semanas. Su prevalencia es difícil de estimar, pero se la considera aproximadamente entre el 4 al 5% y es una causa frecuente de derivación a los médicos especialistas gastroenterólogos. Las alteraciones en la secreción gástrica, biliar y pancreática, así como los defectos en los mecanismos de la absorción mucosa, bien por la alteración en los transportadores específicos o por una lesión inflamatoria con lesión epitelial, mucosa, transmural, patología linfática o incluso por aumento de presión vascular, producirán diarrea. Conocer los mecanismos fisiopatológicos y las distintas patologías que causan diarrea es necesario para llevar adelante el proceso diagnóstico con un adecuado balance costo-efectividad.
Para el abordaje diagnóstico de los pacientes con diarrea crónica, el primer paso es realizar una detallada historia clínica (HC) que deberá incluir los siguientes datos:
- edad;
- antecedentes familiares: cáncer colorrectal (CCR), enfermedad inflamatoria intestinal (EII) o enfermedad celíaca (EC);
- antecedentes quirúrgicos: gastrectomía, resección intestinal, intestino corto, bypass gástrico, colecistectomía, etc.;
- medicación crónica: magnesio, antihipertensivos, hipoglucemiantes, antibióticos, antiarrítmicos, inmunosupresores, AINEs, antineoplásicos, etcétera;
- medicación en los últimos meses: ATB, inmunosupresores;
- pérdida de peso: peso habitual, peso actual y porcentaje de pérdida de peso;
- presencia de dolor abdominal;
- características de las deposiciones: presencia de moco, pus, sangre o esteatorrea;
- relación de la diarrea con la ingesta, el ayuno y el sueño;
- presencia de incontinencia;
- condiciones clínicas asociadas: DBT, hipo/hipertiroidismo, esclerodermia o radioterapia;
- alimentos desencadenantes de los síntomas: cafeína, alcohol, FODMAP, edulcorantes, lactosa, fructosa, etc.;
- viajes recientes a zonas endémicas.
Todos estos datos nos ayudarán a orientarnos hacia la sospecha de una diarrea “funcional” vs. una diarrea de causa “orgánica” (Tabla 1) y también diferenciar la diarrea de colon vs. del intestino delgado (Tabla 2). El siguiente paso será solicitar laboratorio general, específico o biomarcadores. Más adelante analizaremos la utilidad de las distintas herramientas diagnósticas disponibles. Todos estos datos nos permiten clasificar la diarrea como acuosa, malabsortiva o inflamatoria (Figura 1). Siempre debemos considerar la diarrea ficticia o por uso de laxantes y descartar la incontinencia (en pacientes añosos, con antecedentes de un trauma obstétrico, una cirugía pelviana, DBT o STROKE). El examen físico proporciona datos orientativos frente a la presencia de la hipotrofia muscular, edemas, repercusión del estado nutricional, palidez cutáneo-mucosa, lesiones en la piel y mucosas por hipovitaminosis (aftas orales, queilitis, etc.), o lesiones dermatológicas específicas (eritema nodoso, pioderma gangrenoso, etc.). El examen abdominal es importante para descartar la presencia de distensión, masas palpables, visceromegalias o adenopatías. El examen proctológico será de suma importancia para detectar la presencia de fístulas, abscesos, pólipos o tumores del recto, evaluar la incontinencia y descartar un prolapso o la impactación fecal. Otras evaluaciones serán de ayuda complementaria como la manometría anorrectal y la ecografía endoanal.
Tabla 1. Diagnóstico diferencial entre la diarrea funcional y la orgánica
Tabla 2. Diagnóstico diferencial entre la diarrea de colon y la del intestino delgado

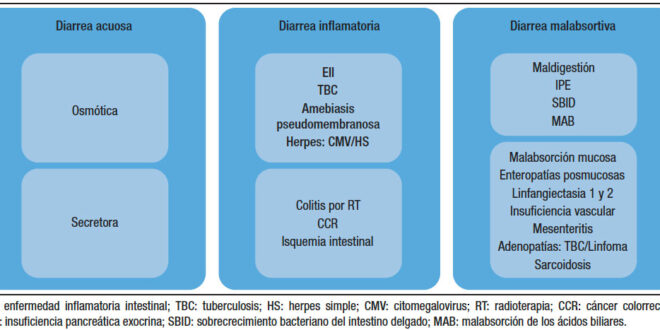
Figura 1. Diarrea acuosa osmótica y secretora; inflamatoria y malabsortiva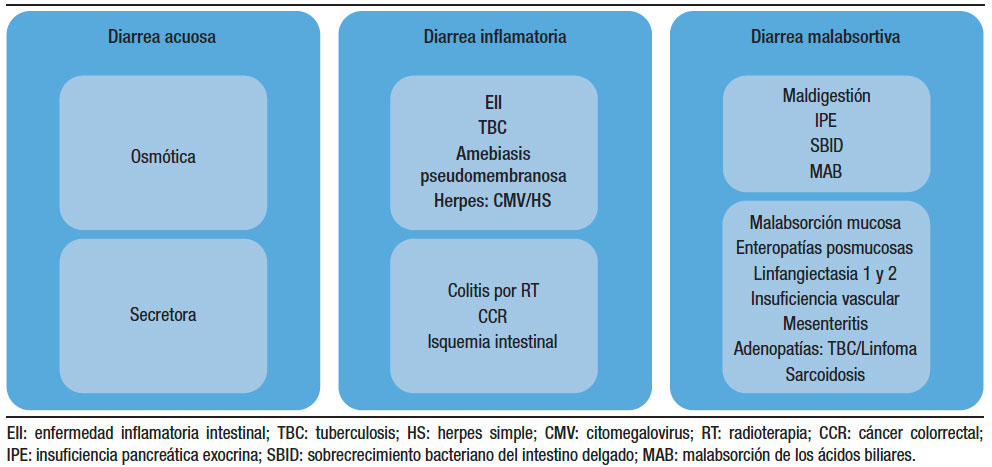
Antecedentes quirúrgicos: debemos tener en cuenta que las resecciones extensas de íleon y colon derecho pueden producir diarrea. Las causas son múltiples: incremento en la velocidad del tránsito intestinal por pérdida de la válvula ileocecal (VIC) y de las hormonas sintetizadas en el íleon terminal, que normalmente regulan el tiempo de tránsito; síndrome del intestino corto y déficit de sales biliares (SB). El íleon es el sector específico de reabsorción de las SB y las resecciones ileales extensas (mayores a 100 cm) se acompañan de la disminución de la cantidad de las SB disponibles para llegar a una concentración micelar crítica necesaria para la correcta absorción de las grasas, pudiendo aparecer esteatorrea. Las resecciones más cortas del íleon (menos de 100 cm) pueden provocar diarrea colerética por la acción de las SB sobre el colon en continuidad. También puede desarrollarse el sobrecrecimiento bacteriano del intestino delgado (SBID). Otras situaciones son: la diarrea colerética poscolecistectomía (hasta en un 10% de los pacientes) y las gastrectomías Billroth II con un asa aferente muy larga que pueden ser una fuente del SBID. Por otro lado, el vaciamiento gástrico acelerado posgastrectomía justifica la aparición de la diarrea que puede, aunque no siempre, asociarse a los síntomas de hipovolemia, hipotensión, palpitaciones y flushing (síndrome de dumping). Las resecciones masivas del intestino delgado, debidas generalmente a la enfermedad de Crohn, la isquemia mesentérica o la enterocolitis necrotizante, pueden provocar una diarrea crónica en el contexto de un síndrome de intestino corto.2
Laboratorio general: la evaluación inicial de un paciente con diarrea crónica deberá incluir una analítica que incluya un hemograma completo, la eritrosedimentación (ESD), la proteína C reactiva (PCR), los electrolitos séricos (sodio, potasio, calcio, magnesio y fósforo), la función renal y el hepatograma, el tiempo de protrombina, las proteínas totales, la albúmina, el colesterol, la función tiroidea y el proteinograma electroforético. Estas pruebas proporcionan información acerca del impacto de la diarrea sobre el estado general y nutricional del paciente y, a menudo, ofrecen pistas importantes para el diagnóstico. La existencia de anemia, trombocitosis, ESD y PCR elevadas, hipoproteinemia, hipoprotrombinemia o hipocolesterolemia constituyen claros indicadores de una enfermedad de base orgánica. El hemograma aportará datos como la presencia de la anemia (ferropénica o megaloblástica) y la eosinofilia (parasitosis, alergias, gastroenteritis eosinofílica). La hipoalbuminemia tiene alta especificidad para la presencia de una enfermedad orgánica (enteropatía perdedora de proteínas, EII, desnutrición). La hipogammaglobulinemia podría orientar a inmunodeficiencias y la hipergammaglobulinemia a procesos inflamatorios o infecciosos (descartar siempre VIH). La presencia de la deficiencia de hierro es un indicador sensible de la patología del intestino delgado, particularmente la EC. Ante sospecha de la EC se solicitará la serología específica: anticuerpos antitransglutaminasa IgA (a-tTG IgA) + IgA total o a-tTG IgA/DGP IgG y anticuerpos antiendomisio (EmA IgA) en casos dudosos, como prueba confirmatoria. La prueba serológica para el virus de la inmunodeficiencia humana (VIH) ayudará a la investigación en los pacientes positivos para descartar causas frecuentes de diarrea en este grupo, como las infecciones oportunistas (Cryptosporidium, Microsporidium, Isospora belli y norovirus) o la enteropatía crónica por enfermedad del SIDA. El TSOMF será de utilidad si los síntomas son sospechosos de malignidad, ante la ausencia de sangrado rectal evidente. La calprotectina fecal (CPF) (si < 40 años) es útil en el diagnóstico diferencial del SII y EII en los adultos en quienes no se sospecha cáncer. La CPF es una proteína que se encuentra en los neutrófilos y, en presencia de inflamación intestinal activa, los neutrófilos migran a la mucosa intestinal. Cualquier alteración de la arquitectura de la mucosa, debido al proceso inflamatorio, da como resultado la fuga de los neutrófilos y, por lo tanto, de la calprotectina en la luz y su posterior excreción en las heces. Se utiliza como nivel de corte el valor de 50 μg/g. Otras causas de la CPF elevada incluyen el CCR, la gastroenteritis infecciosa y las drogas como los AINEs y los IBP. Los niveles mayores a 250 μg/g de heces sugieren una inflamación activa que se correlaciona con la inflamación endoscópica. Un corte de 50 μg/g mejora la sensibilidad, pero disminuye la especificidad, lo que resulta en investigaciones invasivas innecesarias. Sin embargo, un corte de 250 μg/g puede mejorar la especificidad, pero perder o retrasar el diagnóstico de la EII en varios pacientes.3
Estudio de la materia fecal: cálculo del anión gap (AG). Para discernir entre las diarreas osmóticas o las secretoras, se solicitará un ionograma fecal. Se calcula con la siguiente fórmula: 290 – [2 x (Na+ + K+) MF]. En la diarrea osmótica el AG será mayor a 125 mmol/L y en la secretora, menor de 50 mmol/L. Muchas veces los fenómenos que generan la diarrea son mixtos y los resultados serán sólo orientativos. Otros: volumen de materia fecal en 24 horas, pérdida de grasa (pruebas de Van de Kamer o esteatocrito) y clearance de α1 antitripsina (aumentado en enteropatía perdedora de proteínas, como en la EII, yeyunoileitis ulcerativa y linfangiectasia intestinal). La infección por Clostridium difficile debe sospecharse en los pacientes con antecedentes del uso de antibióticos, pero también en grupos de mayor riesgo (IRC, DBT, inmunosuprimidos, trasplantados, embarazadas y en recaída de pacientes con la EII). El diagnóstico se podrá realizar en dos etapas. El primer paso identifica la presencia del organismo con determinación de la enzima glutamato deshidrogenasa (GDH por método de EIA) o pruebas de amplificación de los ácidos nucleicos (PCR). La segunda etapa busca demostrar la producción activa de la toxina C. difficile (toxina A y B por método de EIA). Tal combinación, de una prueba sensible y luego una prueba específica, da altos valores predictivos negativos y positivos cuando las pruebas concuerdan. Determinación de pH fecal: la malabsorción de los carbohidratos resultará en un pH < 6, ya que a medida que los carbohidratos alcanzan el colon son fermentados por las bacterias, liberando ácidos grasos de cadena corta (AGCC) que acidifican las heces.1, 2
Diarrea funcional. Síndrome de intestino irritable
El SII afecta a aproximadamente el 20% de la población. Los síntomas del SII-D y la DF muchas veces se asemejan a los presentes en otras situaciones clínicas como en la EC, el SBID, la intolerancia a los hidratos, la intolerancia a los FODMAP y la sensibilidad al gluten no celiaca (SGNC). El diagnóstico diferencial es un desafío y las distintas pruebas diagnósticas y terapéuticas serán de gran ayuda. Es importante evaluar la presencia de los signos y los síntomas de alarma, llamados “banderas rojas”: la pérdida de peso no intencional, el sangrado rectal, la historia familiar de cáncer colorrectal, la anemia, la masa rectal o abdominal, los síntomas nocturnos, los marcadores de inflamación elevados (ESD o PCR), la diarrea sanguinolenta o mal estado general y la diarrea de < 3 meses de duración nos orientan a causas orgánicas. La ausencia de ellos, en conjunto con síntomas como los definidos en los criterios de Roma IV (Tabla 3) y un examen físico normal, sugiere un trastorno intestinal funcional: un síndrome del intestino irritable con predominio de diarrea (SII-D) o una diarrea funcional (DF). Su especificidad es solo de aproximadamente del 52 al 74% y estos criterios no excluyen de manera confiable la EII, la colitis microscópica o la diarrea por ácidos biliares.4
Tabla 3. Criterios de Roma IV para el diagnóstico del SII

Enteritis actínica
La enteritis actínica (EA) ocurre en hasta el 20% de los pacientes tratados con irradiación pélvica, típicamente 1,5 a 6 años después de la irradiación, aunque son posibles presentaciones posteriores. Los factores de riesgo incluyen: la cirugía abdominal previa, ciertas comorbilidades, la dosis de radiación, el fraccionamiento y la técnica, como así también, la quimioterapia concomitante. Es causada por el daño directo a los enterocitos y la isquemia por daño de los vasos sanguíneos. La fibrosis y el daño linfático son comúnmente vistos. Los pacientes pueden presentar cuadros de diarrea crónica como consecuencia del daño intestinal. El intestino dañado pierde capacidad de absorción y está predispuesto al SBID, particularmente si se desarrollan estenosis. Si está implicado el íleon distal se podrá presentar una malabsorción de los ácidos biliares (MAB).1, 2
Diarrea crónica acuosa osmótica
Se debe a la ingesta de sustancias poco o no absorbibles y osmóticamente activas que atraen agua hacia la luz intestinal, suele ser de volúmenes no mayores a 1 litro y cede con el ayuno. Solemos denominar reacciones adversas a los alimentos como causas de la diarrea osmótica.
Causas:
- Malabsorción de carbohidratos: déficit de disacaridasas. Intolerancia a lactosa y fructosa.
- Ingesta de antiácidos con magnesio.
- Ingesta de laxantes osmóticos (lactulosa, polietilenglicol, fosfatos, sulfato).
- Ingesta excesiva de carbohidratos poco absorbibles: lactulosa; sorbitol, manitol (presentes en golosinas y chicles “sin azúcar”), fructosa; fibra.
- Síndrome de la malabsorción.
Reacciones adversas a los alimentos
Como causa frecuente de la diarrea osmótica, debemos tener en cuenta las reacciones adversas a los alimentos. Denominamos reacciones adversas a los alimentos a la aparición de síntomas a partir de reacciones a diferentes componentes de los alimentos. Incluyen: 1) las mediadas inmunológicamente, como la alergia alimentaria y la EC, que son provocadas por proteínas alimentarias y 2) las no mediadas inmunológicamente como la intolerancia a los hidratos de carbono (intolerancia a la lactosa, fructosa, sorbitol, FODMAP, etc.) (Figura 2). Las intolerancias a los hidratos pueden deberse a defectos enzimáticos (ej.: intolerancia a la lactosa), a trastornos a nivel del transporte (ej.: intolerancia a la fructosa) y otros mecanismos que se mencionan a continuación. Los síntomas digestivos que se producen frente a la malabsorción de los hidratos de carbono se deben a los efectos osmóticos que ellos generan atrayendo agua hacia la luz intestinal y generando diarrea osmótica por un lado y, por otro lado, debido a la fermentación de los mismos por las bacterias intestinales, con la consiguiente producción de gas, distensión, meteorismo y dolor abdominal. Los síntomas extraintestinales (cefalea, vértigo, deterioro de la memoria y letargo) se han descrito en hasta el 20% de los pacientes. Estos síntomas sistémicos podrían ser el resultado de metabolitos tóxicos, producidos por la fermentación bacteriana del colon.
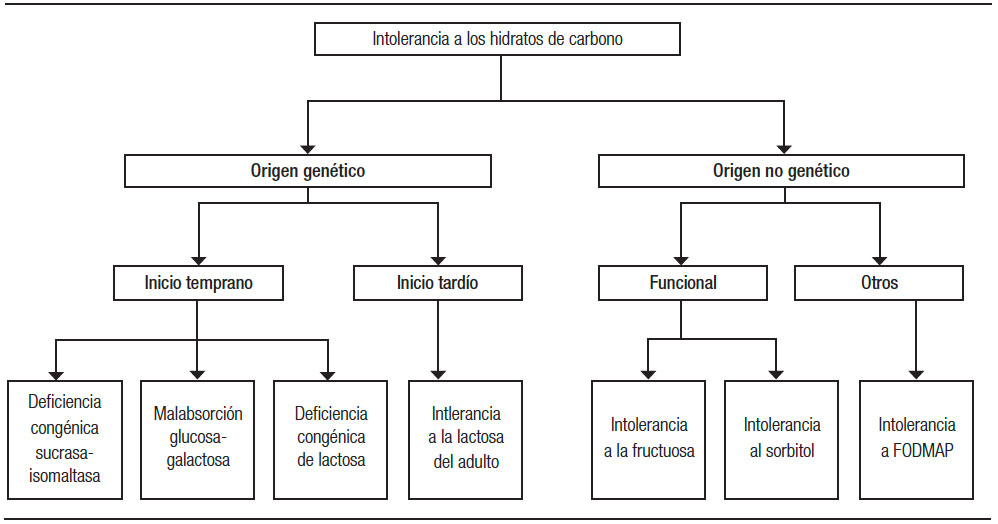
Las intolerancias a los hidratos de carbono pueden dividirse en las de origen genético o de origen no genético (Figura 3). Dentro de las de origen genético se encuentran las de inicio temprano (deficiencia congénita de sucrasa-isomaltasa, de lactasa y la malabsorción glucosa-galactosa) y las tardías como la intolerancia a la lactosa del adulto. Las de origen no genético incluyen las causas funcionales, como la intolerancia a la fructosa, al sorbitol y a los FODMAP.
Figura 2. Reacciones adversas a los alimentos
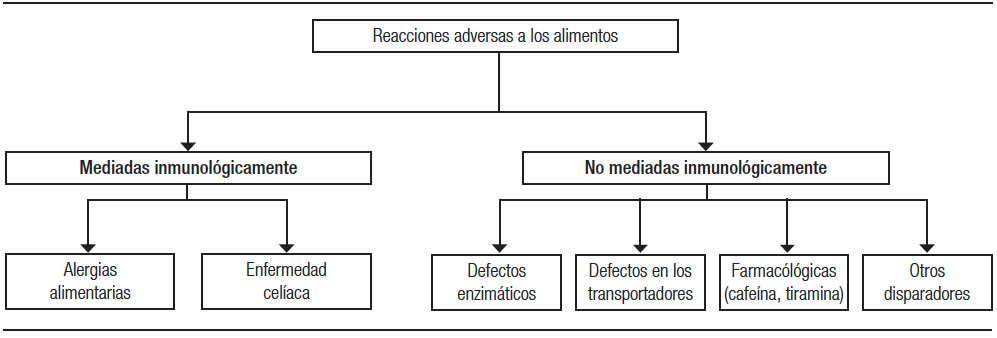
Figura 3. Intolerancia a los hidratos de carbono
Intolerancia a la lactosa: (IL)
Existen tres tipos diferentes:
- deficiencia congénita de lactasa (DCL o agalactia),
- deficiencia primaria, y
- deficiencia secundaria.
1. La DCL es una enfermedad extremadamente rara, autosómica recesiva (mutación en el gen de la lactasa hidrolasa ubicado en el cromosoma 2q21.3) caracterizada por la ausencia o reducción de la actividad enzimática desde el nacimiento. El comienzo de los síntomas (diarrea, falta de crecimiento, alteraciones hidroelectrolíticas) se da en los primeros días después del nacimiento, con el inicio de la lactancia con leche materna o de fórmula que contenga lactosa. Los síntomas desaparecen cuando se modifica la dieta a otra sin lactosa.
2. La intolerancia primaria o hipolactasia es una condición autosómica recesiva frecuente que resulta de un cambio regulado por el desarrollo de la expresión del gen de la lactasa. Es el tipo más común. La malabsorción de lactosa (disacárido) es una condición muy común, caracterizada por la deficiencia de la enzima lactasa (disacaridasa β-galactosidasa) que se encuentra en la membrana del borde en cepillo de la mucosa intestinal. La enzima hidroliza la lactosa en sus componentes galactosa y glucosa en el proceso final de su digestión. Luego, la glucosa y galactosa (monosacáridos) se absorben activamente en el intestino delgado. En los neonatos se encuentran concentraciones fisiológicamente altas de esta enzima. Después del destete, se produce una reducción irreversible y genéticamente programada de su actividad en la mayoría de la población mundial, lo que provoca una malabsorción primaria de lactosa. Cuando la malabsorción de lactosa se asocia con manifestaciones clínicas como la distensión abdominal, el meteorismo, el dolor abdominal y la diarrea, se produce lo que denominamos “intolerancia a la lactosa”. Los síntomas generalmente se desarrollan 30 minutos a 2 horas después de la ingestión de los alimentos que contienen lactosa. En todo el mundo, alrededor del 65% de los adultos pierde la capacidad de digerir la lactosa, con una prevalencia determinada étnicamente que varía desde menos del 10% en los europeos del norte hasta más del 90% en los asiáticos orientales. Pueden experimentar síntomas que comienzan entre los 2 y 3 años o más tarde en la infancia (5-6 años) o adolescencia.5
3. La IL secundaria es una condición transitoria que deriva del daño intestinal secundario a distintas enfermedades como las infecciones, las gastroenteritis virales, la giardiasis, la alergia alimentaria, la EC, el SBID, la enfermedad de Crohn o la enteritis inducida por radiación/quimioterapia. Todas las condiciones patológicas que causan daño mucoso del intestino delgado pueden inducir una reducción de la expresión de la lactasa determinando una deficiencia de lactasa secundaria y transitoria. El tratamiento del trastorno subyacente suele restaurar los niveles de la lactasa y mejorar los signos y síntomas, aunque esto puede llevar tiempo. El diagnóstico de la IL a menudo se realiza sobre una base clínica y la mejoría sintomática surge con una dieta sin lactosa. Sin embargo, hay varios métodos diagnósticos disponibles: la medición de la actividad de la enzima lactasa directamente en las muestras de biopsia de tejido del intestino delgado (procedimiento invasivo y de baja sensibilidad ya que la actividad de la disacaridasa en una pequeña muestra de biopsia no refleja necesariamente la actividad yeyunal en su conjunto). Una prueba genética del polimorfismo C/T13910 Y G/A 22018 puede usarse para la detección de la IL primaria. Los pilares de diagnóstico de la intolerancia a la lactosa de tipo adulto son la anamnesis y el test del aire espirado (TAE) o del aliento con lactosa (prueba rápida y no invasiva) que permite medir el contenido de hidrógeno en el aire espirado. Un aumento del H2 ≥ de 20 partes por millón (ppm) por encima del nivel de referencia, desde el minuto 90 (fermentación por las bacterias colónicas del azúcar no absorbido en el ID), se considera positivo. Resultados falsos negativo o positivo pueden ser secundarios a distintos factores como: las condiciones que afectan la microbiota intestinal (por ejemplo, uso reciente de antibióticos), la ausencia de las bacterias productoras de hidrógeno (10-15% de la población), la ingestión de dietas altas en fibra antes de la prueba, el SBID o trastornos de la motilidad intestinal. El tratamiento consiste en una dieta sin lactosa junto con suplementos de calcio y vitamina D. Las recomendaciones de calcio son de 700 mg/día para niños de 4 a 9 años y 1300 mg/día para mayores de 10 años. Los datos disponibles sugieren que los adolescentes y adultos pueden ingerir hasta 12 g de lactosa (equivalente a 1 taza de leche o 240 ml) sin presentar síntomas o con síntomas mínimos. Se recomienda el consumo de cantidades tolerables de leche o fraccionada a lo largo del día y alimentos con baja cantidad de lactosa como quesos duros. La terapia de reemplazo enzimático con lactasa microbiana exógena (obtenida de levaduras u hongos) representa una posible estrategia para la deficiencia primaria de lactasa. Es importante considerar que la IL puede ser parte de una intolerancia más amplia como es la intolerancia a los FODMAP. Esto está presente en un alto porcentaje de pacientes con el SII y este grupo no sólo requiere restricción de la ingesta de lactosa, sino también una dieta baja en los FODMAP para mejorar los síntomas gastrointestinales.5
Intolerancia a la fructosa
La fructosa es un monosacárido (C6H12O6) que se absorbe a través de un transportador por difusión facilitada. Los transportadores son miembros de la familia de transportadores de glucosa (GLUT). La fructosa se encuentra en frutas, miel y también en el jarabe de maíz de alta fructosa (JMAF) incluido en edulcorantes y refrescos. Se plantea que los transportadores pueden saturarse frente a altas concentraciones de fructosa; es decir que cuando se consume cantidad suficiente como para superar la capacidad de absorción puede producirse diarrea y otros síntomas. Es fundamental la educación alimentaria de los pacientes para evaluar la tolerancia individual, indicar la eliminación (dieta baja en fructosa < 3-5 g de F/porción) por 2 a 6 semanas y luego la reintroducción. En general los pacientes pueden tolerar 10-15 g de fructosa por día y se recomienda el fraccionamiento a lo largo del día. El diagnóstico puede realizarse con el test de hidrógeno en el aire expirado.5
Intolerancia a los FODMAP
Se ha planteado que existen otros componentes de la dieta como posibles disparadores de los síntomas digestivos, dentro de ellos los llamados FODMAP (oligosacáridos, disacáridos, monosacáridos y polioles fermentables). Son carbohidratos de cadena corta, de pequeño tamaño, pobremente absorbidos, con alta actividad osmótica y rápidamente fermentables por la acción de bacterias que, además, modulan la composición de la microbiota y la síntesis de los ácidos grasos de cadena corta (AGCC: acético, propiónico y butírico). Los FODMAP se absorben poco debido, principalmente, a la ausencia de enzimas luminales capaces de hidrolizar los enlaces glucosídicos existentes en los carbohidratos, a la ausencia o baja actividad de las enzimas del borde en cepillo (ej.: lactasa) o la presencia de transportadores epiteliales de baja capacidad (ej.: fructosa, transportador GLUT-2/GLUT-5). Muchos pacientes informan exacerbaciones de sus síntomas después de la ingestión de determinados alimentos, lo que aumenta el interés por el papel que desempeña la dieta, por ejemplo, en el SII. Específicamente, se ha demostrado que las dietas bajas en los FODMAP ayudan a aliviar los síntomas gastrointestinales en estos pacientes. Es probable que los FODMAP promuevan el malestar intestinal a través de los efectos osmóticos, así como a través del aumento de la producción de gas después de una rápida fermentación por bacterias en el intestino delgado distal y colon. Esto es pertinente para la hipótesis FODMAP que sugiere que son las dosis acumulativas, más que los carbohidratos individuales, los principales responsables de los síntomas en el SII.
Existen diferentes tipos: el GLUT-5 de alta afinidad ubicado en la membrana apical, independiente de glucosa y dependiente de un gradiente de concentración; el GLUT-2 de alta capacidad, cotransportador con fructosa y dependiente de glucosa, ubicado en la membrana basolateral y apical, que también transporta activamente glucosa y galactosa; y el SGLT1 cotransportador sodio/glucosa.
Se ha observado dismotilidad intestinal después de la ingestión de poliol. Los estudios muestran un tiempo de tránsito intestinal acelerado, lo que evidencia de forma indirecta el efecto de los polioles sobre la motilidad en el SII. Se presume que esto es secundario a los efectos de la ósmosis y la retención de agua dentro de la luz del intestino delgado, que al aumentar la biomasa podría aumentar la peristalsis. Aunque la malabsorción probablemente explica una parte de los síntomas, es probable que otros factores, incluido el tipo de poliol ingerido, el patrón de consumo y la capacidad del colon para reabsorber agua, también estén involucrados en la inducción de los síntomas. Los FODMAP aumentan el volumen de las heces, mejoran la absorción del calcio, modulan la función de inmunidad y disminuyen los niveles del colesterol sérico, triglicéridos y fosfolípidos. Por otro lado, estimulan selectivamente el crecimiento de algunos grupos microbianos como las bifidobacterias (efecto prebiótico). La fermentación de los carbohidratos pequeños en el colon resulta en la producción de los AGCC que tienen un efecto trófico sobre el metabolismo de los colonocitos al aumentar la producción de energía y la proliferación celular. La dieta baja en los FODMAP mejora los síntomas gastrointestinales globales del SII en el 68-86%, pero pueden tener efectos negativos a largo plazo a nivel nutricional y en el impacto sobre la microbiota intestinal. Con una dieta baja en los FODMAP se pierden los efectos beneficiosos de este grupo de alimentos ya que aportan vitaminas y antioxidantes y, además, esta dieta no soluciona todos los problemas ya que la misma no excluye al almidón resistente ni a la fibra, componentes también altamente fermentativos.5
Diarrea acuosa secretora
Normalmente, el intestino delgado y el colon absorben el 99% del líquido procedente no solo de la ingesta, sino de las secreciones endógenas salivares, gástricas, hepatobiliares y pancreáticas (10 litros de fluido por día). La reducción de la absorción de agua en tan solo un 1% de este volumen total, puede ser causa de una diarrea. Normalmente, en el intestino se produce tanto la absorción como la secreción del agua, aunque la primera es cuantitativamente superior. La diarrea puede surgir tanto por una disminución de la absorción, como por un aumento de la secreción. Este tipo de diarrea, debida a la alteración del transporte epitelial de electrolitos y agua, se conoce como diarrea secretora. Diversas enfermedades afectan el transporte hidroelectrolítico a través de una alteración de los sistemas reguladores, inducida por el efecto de los mediadores de la inflamación (citoquinas, hormonas y toxinas). Todo ello justifica que la lista de las condiciones clínicas asociadas a la diarrea secretora sea muy amplia. Una misma entidad, además, es capaz de producir diarrea por múltiples mecanismos fisiopatológicos: un aumento de la secreción hidroelectrolítica y de la motilidad intestinal, los efectos de sustancias liberadas por las células neuroendocrinas (paracrinas), citoquinas liberadas por células inmunológicas activadas localmente o a distancia, péptidos y hormonas liberados de forma periférica y la propia actividad del sistema nervioso entérico. Estos moduladores pueden actuar, además, sobre más de un efector (función epitelial, contracción muscular, vías para celulares, etc.), ocasionando alteraciones simultáneas en el transporte de los iones, la motilidad o la permeabilidad mucosa. Por otro lado, en cualquiera de estas entidades que causan secreción, la diarrea puede agravarse por la ingesta de sustancias osmóticas. Un ejemplo es la EC donde la diarrea es secretora por la disminución de la absorción del agua y electrolitos, pero osmótica por la alteración de la absorción de los carbohidratos (monosacáridos y disacáridos), sumado a alteraciones de la motilidad gastrointestinal también descritos en esta enfermedad. Las diarreas de causa secretora suelen ser de altos volúmenes (> 1 L) y no ceden con el ayuno.
Causas:
- malabsorción de ácidos biliares (MAB);
- colitis microscópica (CM): colágena (CC) y linfocítica (CL);
- vasculitis;
- abuso de laxantes estimulantes (bisacodilo, antraquinonas-sen/ cáscara sagrada, fenolftaleína);
- fármacos;
- alergias alimentarias;
- alteraciones de la motilidad: diarrea posvagotomía/postsimpatectomía, neuropatía autónoma diabética, SII, alcoholismo;
- causa endocrinológica: enfermedad de Addison, hipertiroidismo;
- TNE: gastrinoma; vipoma; somatostatinoma; síndrome carcinoide, feocromocitoma, carcinoma medular de tiroides, síndrome de Zollinger-Ellison;
- mastocitosis sistémica;
- adenoma velloso de recto.
Colitis microscópica
La CM típicamente se presenta como una diarrea acuosa intermitente que puede estar acompañada de síntomas como el dolor abdominal, la pérdida de peso y la incontinencia. Incluye dos entidades: la colitis linfocítica (CL) y la colitis colágena (CC). Comúnmente se presenta en los pacientes mayores de 65 años y es más frecuente en las mujeres. Se estima que el 10% de las pacientes con diarrea acuosa crónica puede tener una CM. La fisiopatología de la diarrea se debe probablemente a cambios en la mucosa inflamada que llevan a la reducción de la absorción del cloro y sodio, a la inhibición de los canales de cloro/bicarbonato y a la alteración en la permeabilidad. Se asocia a factores de riesgo independientes como el uso de medicamentos (AINEs, IBP, antidepresivos IRS, estatinas y betabloqueantes); el tabaco y condiciones autoinmunes (EC, DBT tipo I, tiroiditis autoinmune y AR). El diagnóstico será definitivo con la observación por parte del patólogo de la presencia de un incremento de linfocitos intraepiteliales > 20/100 células epiteliales en la CL o de una banda de colágeno subepitelial > 10 µm en la CC. La banda de colágeno aparece extremadamente eosinofílica con tinción de rutina H&E, pero es mejor reconocida con la tinción tricrómica de Masson. En el colon sano, la banda de colágeno subepitelial no supera los 3 μm. Es de fundamental importancia realizar la VCC y la toma de biopsias del colon derecho, izquierdo y recto, aún ante la apariencia endoscópica normal de la mucosa en estos pacientes. Se demostró una afectación exclusiva del colon derecho en el 23% de los pacientes, pudiendo no ser diagnosticados con solo una RSC izquierda. El manejo terapéutico incluye: suspender el tabaco y los medicamentos posiblemente asociados, por un lado, y, por el otro, budesonida vía oral, como primera línea de tratamiento a 9 mg/d. Se pueden usar agentes antidiarreicos tradicionales (loperamida, difenoxilato) en casos leves. Como terapia de segunda línea pueden ser utilizados el subsalicilato de bismuto y la mesalazina con o sin colestiramina. La asociación mesalazina/ colestiramina es fisiológicamente razonable por la evidencia previa acerca de la asociación entre la CM y la MAB. La budesonida demostró ser superior a la mesalazina y al placebo en lograr la remisión clínica de los pacientes con la CM y es la droga de elección para el tratamiento de los casos severos. En general, no es necesario realizar una colonoscopia de control para evaluar la respuesta histológica. Se puede considerar suspender la budesonida después de 8 semanas de tratamiento. Un tercio de los pacientes permanecerá libre de síntomas a partir de entonces y no necesitará terapia de mantenimiento, mientras que otros presentarán recaídas. En este sentido, 2 estudios clínicos aleatorizados mostraron que la terapia de mantenimiento con budesonida 6 mg/día durante 6 meses disminuyó el riesgo relativo de recaída clínica (RR: 0,34; IC 95% 0,19-0,6). Aunque el mantenimiento con budesonida puede comenzar con 6 mg, en la práctica, comúnmente se utiliza la mínima dosis efectiva. Se puede considerar el cese de la terapia de mantenimiento después de 6 a 12 meses. Aunque la biodisponibilidad sistémica de la budesonida es baja, el uso prolongado puede predisponer a osteoporosis. Terapias alternativas propuestas en casos refractarios son: metotrexato, anti-TNF y otros inmunomoduladores.6-9
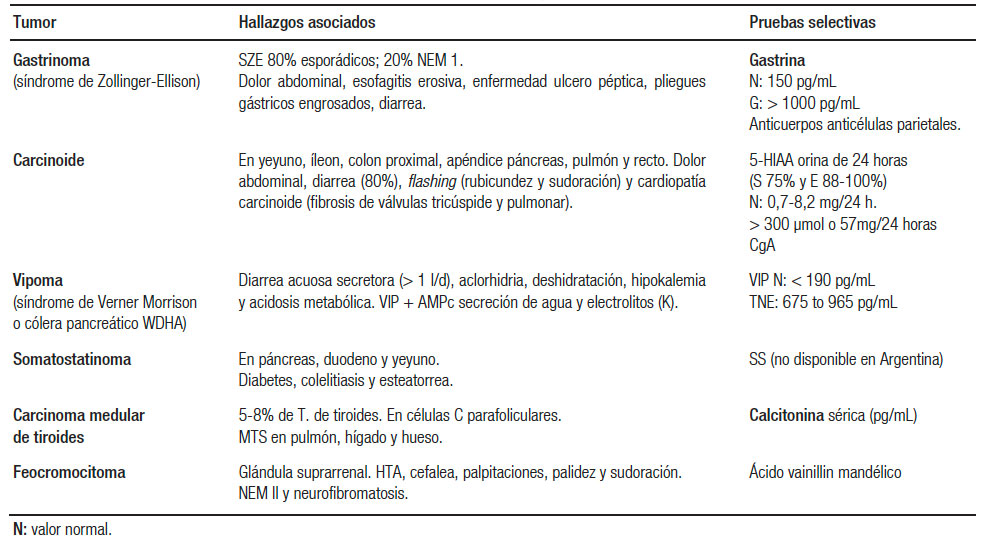
Tumores neuroendocrinos (TNE)
Los TNE representan < 1% de las causas de diarrea crónica, pero deben tenerse en cuenta en el diagnóstico diferencial de las diarreas secretoras. Ante una diarrea acuosa profusa, con volúmenes fecales superiores a un litro diario, frecuentemente asociados a la hipopotasemia, la deshidratación y, en ocasiones, la acidosis metabólica, debe sospecharse la presencia de un TNE secretor de hormonas. Un 25-30% de los TNE-GEP es funcionante. Los TNE funcionantes secretores de péptidos son raros, pero frente a la sospecha deben solicitarse los estudios por imágenes convencionales y funcionales (la TC con contraste endovenoso dinámico, RM, entero TC o entero RM con contraste, PET/TC con 68Galio, 111In/99mTc-Octréotido SPECT/TC). Los TNE son generalmente lesiones hipervascularizadas (realce arterial). En cuanto a las lesiones hepáticas, el patrón de realce permite identificar lesiones metastásicas de origen neuroendocrino y diferenciarlas de las secundarias a adenocarcinoma y otros tumores. La determinación de péptidos en suero (gastrina; serotonina; cromogranina A; histamina; glucagón; cortisol; VIP; calcitonina) o en orina (ácido 5-hidroxiindolacético; ácido vanilmandélico/metanefrinas; histamina) ayudará al estudio funcional ya que se consideran marcadores tumorales. La cromogranina A (CgA) es una glicoproteína que suele estar presente en todas las células endocrinas y neuroendocrinas y es considerada un marcador tumoral sensible para los TNE (sensibilidad 83% y especificidad 96%). Es útil como biomarcador en todos los TNE bien diferenciados independientemente de su localización, tanto en los funcionantes como en los no funcionantes. Es un buen marcador para evaluar la respuesta terapéutica e identificar recidiva y señala el crecimiento de la masa tumoral, por lo que es de utilidad pronóstica. Los análogos de somatostatina (SST), octreótido y lanreótido demostraron eficacia en el control de los síntomas en los tumores funcionantes con el síndrome carcinoide (diarrea severa y flushing) con dosis de 20 y 30 mg cada 28 días respectivamente. En la tabla 4 se resumen las principales características clínicas, localización y marcadores tumorales específicos de los TNE funcionantes que pueden presentar diarrea.
Tabla 4. Características principales de los TNE-GEP funcionantes que pueden presentar diarrea
Diarrea malabsortiva
La malabsorción suele ir acompañada de esteatorrea y deposiciones voluminosas, malolientes y de color claro amarillento. Sin embargo, formas más leves de malabsorción pueden no dar lugar a ninguna anormalidad en las heces. Consideramos maldigestión a la alteración en la hidrólisis de los nutrientes que sucede a nivel de la luz intestinal. Debemos pensar como posibles causas: la insuficiencia pancreática exocrina (IPE), el sobrecrecimiento bacteriano del intestino delgado (SBID) y la malabsorción o concentración inadecuada de los ácidos biliares (MAB).
Insuficiencia pancreática exocrina (IPE)
Es producida por numerosas patologías pancreáticas y extrapancreáticas frecuentes en la práctica clínica habitual. Es el estado de mal digestión de nutrientes causado por la insuficiente secreción de las enzimas pancreáticas, su inadecuada activación, su degradación precoz o la ineficiente interacción enzima-alimento, lo cual condiciona una malnutrición calórico-proteica y el déficit de vitaminas que impactan negativamente en la morbimortalidad de los pacientes, ya que los predispone a padecer complicaciones cardiovasculares, infecciones y fracturas óseas. Puede deberse a la reducción de las enzimas funcionantes que llegan al intestino, la inactivación de las mismas por un pH inapropiadamente bajo (ej: Síndrome de Zollinger Ellison) y por la alteración en la mezcla con la comida (asincronía después del bypass gástrico). Debemos sospecharla en los pacientes con antecedentes de alcoholismo, pancreatitis aguda necrotizante, pancreatitis crónica, fibrosis quística, resección pancreática y cáncer de páncreas, pero también en otras patologías como la DBT (26-44%), la EC (30%), el SII (6%) y la EII (19-30%). Los pacientes pueden presentar distensión y dolor abdominal, meteorismo, diarrea esteatorrea, pérdida de peso, déficit vitamínico (ADEK) y desnutrición. Para el diagnóstico existen pruebas de la función digestiva como el test de Van de Kamer o esteatocrito para confirmar la esteatorrea. El diagnóstico funcional se realizará con la determinación de la elastasa en la materia fecal, una enzima producida y liberada por el páncreas que permanece intacta durante el tránsito intestinal. Son considerados valores normales 200-500 μg/g, con valores de 100-200 μg/g en la IPE leve a moderada y < 100 μg/g en la IPE severa. Es un método altamente sensible y específico para la IPE avanzada (S: 73-100% y E: 80-100%) y su rendimiento es menor para formas leves a moderadas (S < 60%). La prueba no se ve afectada por la terapia concomitante con las enzimas pancreáticas de reemplazo y requiere solo una muestra de heces de 100 mg. Los parámetros bioquímicos a evaluar en estos pacientes son: la albúmina, la transferrina, las vitaminas liposolubles (ADEK), la vitamina B12, el hierro, el zinc, el magnesio, la hemoglobina y la hemoglobina glicosilada. El tratamiento se realiza con las enzimas de reemplazo, el plan de alimentación adecuado y la suspensión de los hábitos tóxicos (especialmente el tabaco y el alcohol). Se recomienda iniciar con 20.000-25.000 UI de lipasa durante las ingestas: 2 cápsulas con el almuerzo y la cena, y 1 cápsula con el desayuno y la merienda. Las dosis podrán aumentarse progresivamente de modo que sean suficientes para lograr el control de los síntomas y la normalización del estado nutricional. Se recomienda suplementar con vitaminas y minerales.11
Sobrecrecimiento bacteriano del intestino delgado (SBID)
La evidencia sugiere que el dolor, la distensión abdominal, la flatulencia y la diarrea son los síntomas más comunes descritos en los pacientes con el SBID. En los casos severos, pueden ocurrir deficiencias nutricionales que incluyen a la vitamina B12, la vitamina D y las deficiencias de hierro, pero, en la mayoría de los casos, estas son sutiles o indetectables. Algunos pacientes también pueden manifestar fatiga y falta de concentración. Sin embargo, ningún síntoma puede atribuirse específicamente al SBID. Los síntomas a menudo se superponen con los de otros diagnósticos como el SII, la DF o la dispepsia funcional. El cultivo del material intestinal, si bien es considerado la prueba de oro diagnóstica, tiene limitaciones: es invasivo, tiene un alto costo, presenta una incapacidad potencial para detectar cepas bacterianas difíciles de cultivar, la detección sólo del SBID es proximal y hay una potencial contaminación de la muestra. La prueba del aliento (método seguro y no invasivo) podrá realizarse utilizando distintos sustratos como la glucosa (G) o la lactulosa (Lac). Ambos sustratos se metabolizan cuando se exponen a las bacterias intestinales, con la producción del hidrógeno (H2) y el metano (CH4). Estos gases son absorbidos desde el tracto gastrointestinal hacia la sangre y, finalmente, exhalados a través de los pulmones. Se considera diagnóstico un aumento en las concentraciones de H2, con respecto al basal > 20 ppm en 90 minutos y de CH4 de > 10 ppm dentro de las 2 horas. La sensibilidad de la prueba de la glucosa en el aliento varió del 20 al 93% y la especificidad del 30 al 86%. La sensibilidad de la prueba de aliento con la lactulosa osciló entre el 31 y el 68% y la especificidad osciló entre el 44 y el 100%. La Lac puede acelerar el tránsito intestinal, dando resultados falsos positivos. Y para la G, una prueba negativa excluye el SBID proximal, pero no el distal ya que la glucosa se absorbe completamente en el yeyuno proximal. Finalmente, podría ser diagnosticado sobre la base de una respuesta clínica clara a un tratamiento empírico con antibióticos. El tratamiento con rifaximina u otros antibióticos de amplio espectro como el metronidazol suele ser efectivo. Algunos pacientes pueden requerir repetir los antibióticos en un nuevo ciclo del tratamiento y será de suma importancia tratar la condición subyacente que permitió el desarrollo del SBID, si es posible (Tabla 5).12, 13
Tabla 5. Factores de riesgo subyacentes para el desarrollo del SBID
Malabsorción de ácidos biliares (MAB)
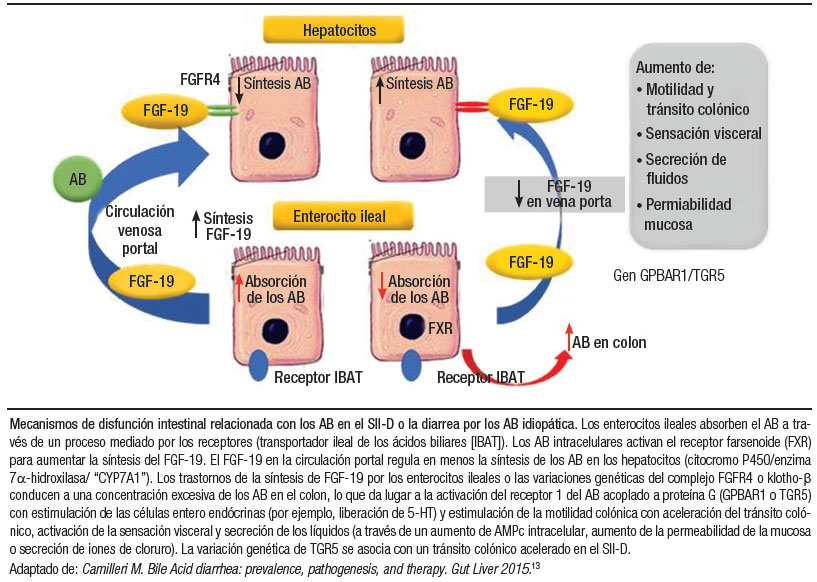
La MAB es una causa muy frecuente de diarrea en los pacientes con la enfermedad de Crohn y el SII-D (Figura 4). De igual modo, la MAB es causa importante y tratable en los pacientes con diarrea idiopática crónica. La prevalencia estimada de la MAB es > 90% en los pacientes con la enfermedad de Crohn con resección ileal y del 11 al 52% en los pacientes no resecados. Es un factor importante en la fisiopatología del SII-D y se encuentra en un tercio de estos pacientes. Además, el 1% de la población general puede tener MAB. Los ácidos biliares (AB) son necesarios para la digestión de las grasas. Se estima que se sintetizan en el hígado 0,5 g de AB/día. La reserva total de los AB es de aproximadamente 3 g, de los cuales el 95% es recuperado a nivel ileal. Este ciclo se repite de 4 a 12 veces/día, lo que da como resultado una pérdida neta en las heces de solo 0,2 a 0,6 g/día (1-3%).18 Los AB primarios, principalmente el ácido cólico (CA) y el ácido quenodesoxicólico (CDCA), se sintetizan en el hígado a partir del colesterol y se secretan a la bilis conjugados con glicina o taurina para aumentan su solubilidad en agua. En el colon, los AB primarios se desconjugan y deshidroxilan a los AB secundarios, predominantemente el ácido desoxicólico (ADC) y el ácido litocólico (ALC) por la acción bacteriana. El colon reabsorbe por difusión pasiva alrededor del 75% de los AB. En el colon, los AGCC y el ADC estimulan la secreción de líquidos y la motilidad, es decir que niveles altos de los AB a nivel colónico provocan la diarrea acuosa. Hasta el 20% del almidón de la dieta escapa a la absorción en el intestino delgado, lo que resulta en la generación de los ácidos grasos de la cadena corta (AGCC) por las bacterias del colon. Se postula que los AGCC estimulan la liberación colónica de la serotonina (5-HT) por las células enteroendócrinas. Las causas de la diarrea por los AB incluyen: una deficiencia en el factor de crecimiento de fibroblastos 19 (FGF-19) (hormona producida en los enterocitos, que regula la síntesis de los AB hepáticos); variaciones genéticas que afectan las proteínas involucradas en la síntesis y circulación enterohepática de los AB o en el receptor TGR5, que media las acciones de los AB (secreción y motilidad) en el colon. El TGR5 es un receptor importante que se expresa en las neuronas entéricas, las células enteroendócrinas y las neuronas espinales primarias aferentes involucradas en la transducción sensorial para mediar los efectos de los AB sobre la motilidad, directamente por su acción en las neuronas e indirectamente al estimular la liberación de serotonina. Los AB aumentan la permeabilidad mucosa, inducen la secreción de agua y electrolitos por los colonocitos (cloruro y el intercambio apical de Cl− / OH−) y aceleran el tránsito colónico probablemente a través de la estimulación de las neuronas ganglionares mientéricas por el TGR5, en parte estimulando las contracciones colónicas propagadas de gran amplitud.15, 16
Figura 4. Circulación enterohepática de los AB. Diarrea por los AB en el SII-D
Se ha clasificado la malabsorción de las sales biliares en tres tipos:
Tipo I: secundaria a resección o enfermedad ileal activa (E. Crohn, TBC, RT). Puede dar diarrea colerética por estimular la secreción de cloro y agua a nivel colónico o esteatorrea por la malabsorción de grasas al disminuir la concentración micelar crítica.
Tipo II o idiopática: asociada con el aumento en la producción. Se estima que ocurre en el 25-28% de los pacientes con la DF y el SII-D basado en un 75SeHCAT < 10%. Se asocia con un defecto en la regulación en menos de la síntesis de los AB mediada por FGF19, llevando a sobreproducción de los AB (por disminución de los niveles circulantes de FGF-19).
Tipo III no relacionada con la disfunción ileal: asociada con otras condiciones gastrointestinales: poscolecistectomía, posvagotomía, la EC, el SBID, la pancreatitis crónica (PC) y la colitis microscópica (CM). Se asocia con tiempos de tránsito intestinal acelerados.
Una cuarta categoría puede resultar de una síntesis hepática excesiva de los AB; por ejemplo, el fármaco hipoglucemiante oral metformina se asocia con un aumento de la síntesis de los AB hepática.13, 18
Diagnóstico: tradicionalmente, se ha argumentado que el patrón oro para el diagnóstico de la diarrea por los AB es una respuesta clínica favorable al tratamiento empírico con un quelante de SB (colestiramina). Sin embargo, estos medicamentos presentan mala palatabilidad, interacciones farmacológicas y efectos secundarios, como el dolor abdominal, las náuseas y la constipación. También requieren una titulación de la dosis para optimizar el beneficio clínico. Por todas estas razones, su uso puede ser engorroso y la adherencia por parte de los pacientes suele ser baja. Hay varios métodos para diagnosticar la MAB: la prueba de la retención del ácido homotaurocólico marcado con selenio (75SeHCAT, prueba de medicina nuclear), medición de C4 y FGF19 en suero y los AB en las heces. El patrón oro es el 75SeHCAT.
1) Gammagrafía con 75SeHCAT (sensibilidad: 80-94%; especificidad: 70-100%): esta prueba utiliza los AB sintéticos administrados por vía oral, marcados con selenio, que es resistente a la degradación bacteriana y a la absorción pasiva por el intestino delgado. Así, los niveles de selenio identificados por la cámara gamma en el día 1 y 7 proporcionan una evaluación indirecta de la absorción y la excreción fecal de los AB. La retención corporal del radiofármaco parece correlacionarse con la absorción ileal. Se ha establecido como valor de corte < 10%. La retención de 75SeHCAT < 10% sugiere MAB moderada y < 5% MAB grave. Las desventajas de esta prueba incluyen la necesidad de administración de un isótopo radiactivo, de infraestructura (cámara gamma) y de múltiples visitas para completar el estudio. Además, no está actualmente disponible en nuestro medio.
2) Medición de C4 en suero en ayunas (ng/mL): refleja la síntesis hepática y se encuentra aumentado cuando se incrementa la síntesis de los AB. El C4 en ayunas > 35 ng/mL demostró una sensibilidad y especificidad del 87% y el 86% en comparación con la retención de 75SeHCAT < 10%. Esto se traduce en una alta razón de probabilidad negativa del 94% y positiva del 71%. El momento de las mediciones es importante ya que el C4 sérico tiene dos picos, uno al mediodía y otro a las 9:00 p.m.
3) Medición de FGF19 en suero en ayunas (pg/ml, ELISA): prueba disponible comercialmente. Sus niveles reflejan la recaptación de los AB luminales por el transportador del AB ileal. El FGF19 ejerce efectos inhibidores sobre la síntesis hepática de los AB, por lo tanto, la disminución de los niveles de FGF19 se asocia con un aumento de la síntesis hepática. Un nivel en ayunas < 145 pg/mL tiene un valor predictivo positivo y negativo de 61% y 82% respectivamente para la MAB, utilizando una retención de 75SeHCAT de < 10% como patrón de oro. Aunque estas pruebas en suero son relativamente simples de realizar, a diferencia de SeHCAT, no están ampliamente disponibles.
4) Medición de los AB fecales en la recolección de 48 horas: se realiza durante los dos últimos días de una dieta alta en grasas y es el estándar de oro para medir la excreción de los AB con un valor > 2,337 μmol/48 horas usado como índice de la MAB. Los AB fecales pueden cuantificarse mediante una cromatografía por cualquiera de los siguientes métodos: cromatografía de gases o líquida de alta resolución o espectrometría de masas. Desafortunadamente, estos métodos llevan mucho tiempo, son complejos, caros y no están siempre disponibles. Además, hay muy pocos estudios que hayan evaluado el rendimiento de estas pruebas.
Se ha observado que existe una mayor proporción de los AB primarios en las heces de los pacientes con el SII-D y se han descrito cambios en el microbioma fecal, inducidos por el perfil de los AB. En el SII-D existen variantes genéticas en la vía FGF19-FGFR4-Klothoβ que afectan al tiempo de tránsito colónico y, por otro lado, los AGCC en muestras fecales de los pacientes con el SII-D se caracterizan por una cantidad más baja de acetato y propionato y mayor de n-butirato (el cual induce secreción). La microbiota intestinal también regula la expresión del FGF 19 en el íleon y la CYP7A1 en el hígado mediante mecanismos dependientes de FXR. La composición de los AB parece ser el principal regulador de la estructura del microbioma. Múltiples estudios de investigación en curso buscan desentrañar las contribuciones del microbioma y la composición de los AB en las distintas afecciones, incluido el CCR, la EII y el SII. Por otro lado, existe un interés renovado en el papel de los AB en otras enfermedades como la obesidad, la diabetes y la infección recurrente por C. difficile.14
Malabsorción
La malabsorción se define como la alteración en la absorción mucosa de los nutrientes. Las enfermedades del intestino delgado son causa común, a menudo subestimada, de diarrea. Muchas enfermedades producen daño en la mucosa intestinal alterando sus funciones digestivas y absortivas, a menudo por el desarrollo de atrofia de las vellosidades intestinales (AV). La malabsorción puede llevar a estados de malnutrición y deficiencias nutricionales. Para el estudio de causas mucosas será necesaria la realización de una video endoscopía digestiva alta y la toma de biopsias para definir la etiología de la enteropatía, ya sea, una enfermedad celíaca o cualquier otra entidad que cursa con atrofia vellositaria (Tabla 3). Para las enteropatías segmentarias o distales (E. Crohn, TBC, linfoma o yeyunoileítis ulcerativa) serán de utilidad los estudios por imágenes como la entero TC o entero resonancia, la video cápsula endoscópica y las enteroscopías dirigidas con toma de biopsias. También la tomografía y la RM con contraste serán de utilidad para el diagnóstico de las patologías con compromiso posmucoso (linfangiectasia intestinal), mesentérico, ascitis, adenomegalias y EII.
Causas de diarrea por malabsorción
1. Enfermedades de la mucosa:
- enfermedad celíaca (EC);
- inmunodeficiencia común variable (IDCV);
- giardiasis/isosporidiasis/criptosporidiasis;
- enfermedad de Whipple (EW);
- enteropatía por drogas (olmesartán, metotrexato, micofenolato, etc.);
- enteropatía en VIH;
- enteropatía autoinmune (EAI);
- esprúe tropical;
- TBC;
- SBID;
- enfermedad de Crohn;
- esprúe colágeno;
- linfoma;
- enfermedad injerto contra huésped.
2. Síndrome del intestino corto
3. Isquemia mesentérica crónica
Diarrea secundaria a las enfermedades del intestino delgado
Enfermedad celíaca
Afecta al 1% de la población con diferencias según la localización geográfica. El riesgo de padecer la EC es mucho mayor en familiares de primer grado (5-10%), así como en los pacientes con DBT mellitus tipo 1 (T1DM) y otras enfermedades autoinmunes asociadas o síndrome de Down, entre otros grupos de alto riesgo. La EC se debe a una reacción inmune al gluten dietario presente en el trigo, la cebada, el centeno y la avena que produce la AV y la malabsorción. Es considerada una enfermedad sistémica, autoinmune, inmunomediada y con factores genéticos predisponentes. Existe un gran espectro de manifestaciones clínicas, desde el clásico cuadro de la malabsorción con diarrea, la distensión abdominal, la pérdida de peso y el déficit vitamínico; síntomas similares a un SII, hasta las formas de presentación atípicas o con síntomas extraintestinales (anemia ferropénica, osteoporosis, hipertransaminasemia idiopática, infertilidad y síndromes neurológicos). Pueden presentarse manifestaciones clínicamente graves en el posparto, especialmente durante el puerperio en el 15-20% de las mujeres celíacas. El diagnóstico se basa en las pruebas serológicas específicas de alta sensibilidad y especificidad. La prueba inicial de elección es el anticuerpo antitransglutaminasa (anti-tTG) tipo IgA en personas mayores de dos años de edad y la determinación concomitante del nivel de IgA Total. La deficiencia de IgA ocurre en el 2% de los pacientes con una EC y se asocia con frecuencia a otras enfermedades que pueden causar AV, como la giardiasis, el SBID o la IDCV. En los individuos con la deficiencia de IgA confirmada deben realizarse pruebas serológicas (anticuerpos) de tipo IgG, tales como los péptidos deaminados de gliadina IgG (DGP- IgG) o la antitransglutaminasa tisular IgG. Los anticuerpos anti-TG2 son los más sensibles para el diagnóstico de la EC, mientras que los anticuerpos antiendomisios (EMA IgA) son los más específicos, pero no siempre están disponibles, tienen un costo mayor, dependen de un operador y, además, requiere la técnica de inmunofluorescencia. Por lo tanto, el anti-TG2 se utiliza como el primer paso y el EMA IgA se puede utilizar como prueba de confirmación, particularmente cuando el TG2 tiene un título bajo o en casos discordantes. La combinación de anti-tTG IgA/DGP IgG es considerada la mejor herramienta para identificar la EC ya que permite el diagnóstico aún en los pacientes con deficiencia de IgA. Los signos endoscópicos que pueden hacer sospechar una AV son: la pérdida o la reducción de los pliegues duodenales, el patrón mucoso de mosaico o patrón micronodular, la configuración en el peinado de los pliegues duodenales, el incremento del patrón vascular, los surcos y las fisuras. Cuando están presentes tienen alta especificidad (E) cercana al 100% y su sensibilidad (S) varía entre el 47 al 100% en distintas publicaciones. La utilización de tecnologías emergentes en endoscopia para el diagnóstico de la EC alcanza una S y E casi del 100% y son de utilidad, cuando están disponibles, para dirigir las biopsias a zonas afectadas, en el caso de compromiso parcheado. Estas son: la técnica de la inmersión en agua (es simple y rápida), la magnificación o zoom, la endoscopía magnificada de alta resolución (i-SCAN-HDWI), las imágenes de banda estrecha o Narrow-Band Imaging (NBI), las cuales mejoran la visualización del patrón microvascular; y también la cromoendoscopía (con índigo carmín o azul de metileno) o Fujinon Intelligent Chromo Endoscopy System (FICE), solas o combinadas. El diagnóstico se confirma con la realización de la biopsia intestinal, con mayor sensibilidad cuando se toman un número suficiente de muestras (4 de la segunda porción duodenal y 2 del bulbo duodenal). Esto se debe a que la enfermedad puede presentar una afectación en los parches y, además, a que la orientación de las mismas puede ser variable. Las biopsias de bulbo permitirán diagnosticar casos de la denominada “EC ultracorta”, presente en el 9 al 13% de los pacientes, donde se describe la AV exclusiva del bulbo. La anatomía patológica clásica de la EC es la presencia de la AV leve, moderada o severa según la relación entre la altura de las vellosidades y la profundidad de las criptas. Otros hallazgos característicos son: el incremento de los linfocitos intraepiteliales (LIE), la hiperplasia de las criptas que se vuelven profundas, muchas veces, ramificadas, con incremento de mitosis; el aumento del infiltrado linfoplasmocitario en la lámina propia y un epitelio de superficie pseudoestratificado con una disminución de las células caliciformes. La biopsia debe realizarse mientras el paciente consume dieta con gluten, ya que la exclusión del gluten de la dieta lleva, en la mayoría de los casos, a la recuperación de la histoarquitectura normal del ID y será un factor confundidor para la etapa diagnóstica.19
Los pacientes con la atrofia vellositaria (AV) y serologías negativas para la enfermedad celíaca (EC) plantean un dilema diagnóstico y terapéutico. Si bien la prevalencia de la EC seronegativa es de 6 a 22%, existe una serie de enfermedades que pueden mimetizar la enteropatía celíaca y deben conocerse para considerarlas en el diagnóstico diferencial, especialmente en los pacientes sin una respuesta clínica ni histológica a la DLG. La AV se encuentra en otras entidades con características similares o distintivas a la EC que vamos a desarrollar. Fundamentalmente, debemos tener en cuenta las siguientes entidades: la inmunodeficiencia común variable (IDCV), la enteropatía por drogas, la enteropatía autoinmune, la giardiasis, la gastroenteritis eosinofílica, el esprúe tropical y la enfermedad de Whipple.
Inmunodeficiencia común variable
Puede presentarse con la diarrea crónica con esteatorrea o enteropatía perdedora de proteínas. El dato de laboratorio que nos orienta a esta enfermedad es la presencia de la hipogammaglobulinemia. El diagnóstico se establece en base a los niveles reducidos de, al menos, dos inmunoglobulinas séricas, IgG e IgA o IgM, y la producción alterada de los anticuerpos específicos en respuesta a la vacunación in vivo o la infección reciente. Generalmente, se presenta después de la pubertad y antes de los 30 años, con alguna evidencia de una distribución bimodal (1 a 5 y 18 a 25 años). La enfermedad probablemente sea el resultado de una desregulación inmunitaria no definida, que conduce a una falla en la diferenciación de las células B con deficiencia de la secreción de inmunoglobulinas. Hasta el 60% de los pacientes desarrolla una diarrea y el 10% una malabsorción idiopática. Pueden presentar: la AV, la diarrea crónica, el síndrome de la malabsorción, una inflamación del intestino delgado o colon similar a la enfermedad de Crohn (EC) o una colitis ulcerativa (CU) (EII-like en el 30%), la anemia perniciosa, la hiperplasia nodular linfoide (20%), el linfoma y el adenocarcinoma gástrico. La diarrea puede ser causada por diferentes organismos infecciosos. Los más frecuentes son la Giardia lamblia, Salmonela, Shigella y Campylobacter, que pueden ser difíciles de detectar. El hallazgo histopatológico característico es la AV con la ausencia de células plasmáticas en el infiltrado de la lámina propia por H&E. Los patrones histológicos muchas veces pueden simular la colitis linfocítica o colágena, la gastritis linfocítica, la enfermedad granulomatosa y la EII. Los pacientes suelen presentar: a) antecedentes de infecciones bacterianas recurrentes, particularmente de las vías respiratorias superiores e inferiores como la sinusitis, la otitis y la bronquitis recurrentes, la neumonía y pueden llegar a desarrollar una severa fibrosis pulmonar y bronquiectasias. Tienen riesgo de infiltración granulomatosa e intersticial, b) mayor incidencia de enfermedades autoinmunes (anemia perniciosa, enfermedad tiroidea, vitíligo, DBT I, psoriasis, LES, AR, artritis reumatoide juvenil, uveítis, PTI, trombocitopenia inmune o anemia hemolítica) y c) predisposición a desarrollar neoplasias (cáncer gástrico y LNH). Su tratamiento requiere un abordaje multidisciplinario que incluye inmunólogos, clínicos y gastroenterólogos. Se utilizan terapias con inmunoglobulinas parenterales o subcutáneas mensuales o con intervalos a determinar, lo que ayuda a prevenir infecciones severas, pero esta medida terapéutica no parece tener efecto sobre las manifestaciones gastrointestinales. El tratamiento de la diarrea suele ser sintomático. Algunos estudios han demostrado mejoría con la terapia con corticoides. Pueden requerir cursos de tratamiento con antibióticos para tratar el SBID y la giardiasis. El tratamiento con budesonida oral con recubrimiento entérico demostró ser eficaz en la enfermedad Crohn-like con afectación ileocolónica. Las enfermedades linfoproliferativas se manifiestan comúnmente como linfadenopatías y esplenomegalia. El seguimiento incluye una endoscopia semestral y una ecografía/TC anual, la búsqueda y el tratamiento de la gastritis asociada a Helicobacter pylori.20, 21
Enteropatías por drogas
Algunos medicamentos pueden ocasionar la AV, imitar una EC y deben ser considerados como causa de la enteropatía seronegativa. Se ha descrito el desarrollo de la AV y otras alteraciones morfológicas en el intestino delgado en los pacientes que reciben el tratamiento crónico con los AINEs, los inmunosupresores (micofenolato, metotrexato, colchicina, azatioprina, tacrolimus, etc.) y también con olmesartán. La duodenitis temprana asociada a los AINEs se caracteriza por la infiltración inespecífica de la lámina propia por neutrófilos y células plasmáticas, acompañadas en algunos casos de la AV leve y la linfocitosis intraepitelial (LIE). En la enfermedad avanzada por los AINEs, pueden aparecer erosiones de la mucosa (a menudo múltiples) e incluso úlceras profundas que pueden provocar una hemorragia y una perforación. La enteropatía inducida por olmesartán es una nueva entidad clínica que debe incluirse en el diagnóstico diferencial de la AV seronegativa. El olmesartán es un bloqueante del receptor de la angiotensina II que se utiliza para el tratamiento de la hipertensión arterial. Su uso induce una diarrea severa en algunos individuos. La edad media de los pacientes afectados es entre 70 y 72 años, con frecuencia asociada a una dosis media de 40 mg/día y un tiempo de tratamiento prolongado de 2 a 3 años. La evaluación de laboratorio generalmente muestra la malabsorción con anemia normocítica normocrómica, la hipoalbuminemia y múltiples anomalías electrolíticas. La enteropatía puede desarrollarse meses o años después del inicio del tratamiento. El 92% de los pacientes presenta la AV parcial o completa y el 61% tiene aumento de los LIEs que oscilan entre el 25 y el 100%. También puede presentar abundante infiltración de la lámina propia por los neutrófilos y los linfocitos con focos de apoptosis de las células de las criptas. Además, el 22% de los casos muestra el depósito de colágeno subepitelial, parecido a lo observado en el esprúe colágeno. Estos pacientes presentan anticuerpos para la EC negativos y falta de respuesta clínica a la DLG. La mejoría clínica e histológica se logra al suspender la droga, con una recuperación histológica completa de la AV. La presencia de HLA-DQ2 puede conferir un mayor riesgo de desarrollar la AV.22-25
El micofenolato es un fármaco inmunosupresor comúnmente utilizado para prevenir el rechazo agudo de los aloinjertos. El daño por el micofenolato generalmente se manifiesta en el colon, pero también puede estar presente en el duodeno. Los hallazgos histológicos son muy similares a los de la enfermedad del injerto contra huésped agudo (GVHD), como el desorden arquitectónico de la cripta, la lámina propia edematosa con inflamación crónica y la dilatación quística de las criptas duodenales. El revestimiento epitelial de las criptas dilatadas a menudo se aplana y muestra un aumento de la apoptosis, así como los neutrófilos luminales y los restos apoptóticos. La presencia de los eosinófilos (más de 15 por 10 campos de alta potencia) está más en consonancia con el daño inducido por el micofenolato, mientras que la presencia de agregados de células y la degeneración de la cripta hipereosinofílica sugiere la GVHD.23
Enteropatía autoinmune
La enteropatía autoinmune (EAI) es una causa rara de diarrea caracterizada por la presencia de la AV total o parcial. A diferencia de la EC, la histología en la EAI muestra la ausencia de células caliciformes y células de Paneth, el LIE menos prominente e infiltrado linfoplasmocitario en la lámina propia. Otros hallazgos histológicos son la linfocitosis profunda de las criptas y el aumento de los cuerpos apoptóticos en las criptas. Se describió por primera vez en niños pequeños, a menudo recién nacidos, que presentaban 1) una diarrea grave con malabsorción, 2) AV completa, y 3) los anticuerpos antienterocitos en suero presentes en el 85% de los pacientes. Por definición, esta condición no responde a ninguna dieta de exclusión alimentaria. El desorden también ocurre en los adultos. Se cree que el sistema inmunológico se dirige a los componentes del enterocito a través de la activación del sistema inmunológico innato y adaptativo. Una característica común es la asociación con otras enfermedades autoinmunes, típicamente enfermedad tiroidea, miastenia gravis, lupus, síndrome de Sjögren, así como deficiencias inmunes que predisponen a las infecciones o parasitosis. Por lo general, los pacientes tienen una diarrea importante, la serología celíaca negativa y una falta de respuesta a la DLG.19
Gastroenteritis eosinofílica (GE Eo)
La GE Eo forma parte del espectro de las enfermedades gastrointestinales eosinofílicas primarias junto con la esofagitis y la colitis eosinofílica. Estos trastornos inflamatorios se caracterizan por la infiltración predominante de eosinófilos en los tejidos, ante la ausencia de causas conocidas de eosinofilia: parasitosis, las drogas o el cáncer. Su incidencia es estimada en 1 a 30/100.000 habitantes con máxima incidencia en la 4ta y 5ta década. Los trastornos alérgicos concomitantes, como el asma, la rinitis, el eccema y las intolerancias a drogas o alimentos están presentes en el 45% al 63% de los casos reportados. Cerca del 70% de los pacientes presenta una historia familiar de atopía. La forma de presentación clínica varía según la localización de la inflamación eosinofílica en la pared intestinal: el compromiso mucoso (el más frecuente) puede manifestarse con náuseas, vómitos, diarrea, anemia, malabsorción o la enteropatía perdedora de proteínas. El compromiso muscular lleva a cuadros de dolor abdominal u obstrucción intestinal y se evidencia un engrosamiento de la pared intestinal en los estudios por imágenes. El compromiso de la serosa se manifiesta con ascitis eosinofílica. El compromiso seroso y muscular pueden presentar un curso clínico intermitente con recaídas y el compromiso mucoso, un curso crónico continuo. El aspecto endoscópico es inespecífico y puede presentarse con: inflamación crónica inespecífica, eritema mucoso, friabilidad, ulceración, nodularidad o puntos blancos. El examen histológico muestra la infiltración mucosa por eosinófilos mayor a 20 eosinófilos/HPF (> 20 Eo/HPF), valor que se ha considerado como umbral para el diagnóstico. La observación de la AV, la hiperplasia de las criptas o los abscesos también son hallazgos comunes. La distribución irregular de la enfermedad requiere múltiples biopsias, al menos 5 o 6, tomadas tanto de la mucosa endoscópicamente anormal como de la normal. La tomografía computada (TC) puede detectar el engrosamiento difuso de los pliegues de la mucosa intestinal, el engrosamiento de la pared, la ascitis y la obstrucción. La ecoendoscopía también es una herramienta útil para evaluar la afectación muscular y subserosa. El 80% de los pacientes presenta una eosinofilia periférica, aunque no se correlaciona con la actividad clínica de la enfermedad, ni es de utilidad en el seguimiento durante o después del tratamiento. El diagnóstico requiere tres criterios: 1) la presencia de los síntomas gastrointestinales; 2) la evidencia histológica de la infiltración eosinofílica en una o más áreas del tracto gastrointestinal; y 3) la exclusión de otras causas de eosinofilia. En este sentido es crucial descartar en el diagnóstico diferencial: las infecciones parasitarias (Strongyloides, Ascaris, etc.), la hipersensibilidad a las drogas (naproxeno, clozapina, rifampicina, enalapril, carbamazepina, sales de oro, tacrolimus o IFN), la vasculitis (síndrome de Churg-Strauss o poliarteritis nodosa), las enfermedades del tejido conectivo (esclerodermia, dermatomiositis o polimiositis), la EII, la EC, el linfoma/leucemia y la mastocitosis; además, el síndrome hipereosinofílico (trastorno mieloproliferativo caracterizado por la eosinofilia periférica idiopática persistente > 1500 Eo/HPF por más de 6 meses y el compromiso de distintos órganos, incluyendo corazón, SNC, piel, pulmones, hígado, riñones y el tracto gastrointestinal). Es importante realizar una evaluación de alergia alimentaria en todos los pacientes con la sospecha de la GE Eo. Las pruebas de alergia cutánea carecen de sensibilidad (alrededor del 40%) y especificidad. El diagnóstico de la GE Eo representa un desafío y es considerado un diagnóstico de exclusión. Serán de utilidad la solicitud de los parasitológicos seriados, ESD, PCR, IgE, ANCA, serología para la EC, biopsias gástricas, duodenales, ileales y de colon eventualmente. El tratamiento consiste en una dieta de exclusión de los posibles alérgenos, con corticoides que son efectivos en el 80% de los casos (prednisona 20 a 40 mg/d por 2 a 6 semanas). Otras terapias utilizadas son la budesónida (9 mg/d), los antileucotrienos: montelukast (inhibidor selectivo de leucotrieno LTD4) solo o en combinación con corticoides para la inducción y el mantenimiento de la remisión; ketotifeno (agente antihistamínico de segunda generación); azatioprina, infliximab (anti-TNF) y otros agentes biológicos (anti IL-5 mepolizumab y anti IgE omalizumab) en casos de refractariedad.26, 27
Enfermedad de Whipple
La EW es una enfermedad sistémica poco frecuente (prevalencia menor a 0,1%), crónica e infecciosa, causada por la bacteria Tropheryma whipplei. Típicamente compromete al intestino delgado causando una severa malabsorción. Puede afectar a otros órganos como el corazón, el sistema nervioso central (SNC), las articulaciones y el sistema vascular. Afecta principalmente a los pacientes de sexo masculino caucásicos con un pico de incidencia entre los 40 a los 50 años. El hallazgo característico en las biopsias intestinales es la presencia de los macrófagos espumosos PAS+ (ácido periódico-Schiff) con inclusiones resistentes a la diastasa, Ziehl-Neelsen negativos infiltrando la lámina propia. Puede desarrollarse clínicamente en tres etapas: artralgias y artritis (migratorias, no deformantes y de grandes articulaciones) que preceden a la enfermedad varios años, astenia y debilidad. La segunda etapa se caracteriza por los síntomas gastrointestinales (la diarrea, la esteatorrea, el dolor abdominal, la pérdida de peso, la malabsorción, la anemia y el déficit de vitaminas). Con el tiempo pueden desarrollar una desnutrición severa, linfadenopatías, fiebre e hiperpigmentación de la piel. Otros síntomas extraintestinales son la hipertensión pulmonar, la insuficiencia cardíaca congestiva, la endocarditis, la poliserositis (pericarditis, derrame pleural y ascitis), la enfermedad ósea metabólica y trastornos neurológicos en el 20 al 40% (demencia, ataxia cerebelosa, miorritmia oculomasticatoria y oculofacial, etc.). El diagnóstico de la EW se realiza mediante biopsias del intestino y la identificación del organismo. Los criterios diagnósticos actuales requieren dos de los tres siguientes: 1) Tinción de PAS+ que muestre macrófagos espumosos en las biopsias de los tejidos afectados. 2) Detección del T. whipplei o del RNAr 16S específico de la bacteria a partir de material mediante PCR 3) Tinción IHQ con anticuerpos contra T. whipplei a partir de muestras intestinales o de otros tejidos. Los estudios de microscopía electrónica pueden identificar bacterias alargadas con pared celular trilaminar y lisosomas característicos en histiocitos intestinales, pero no siempre está disponible en todos los centros. El diagnóstico diferencial incluye: la EC, la TBC y la infección por Mycobacterium avium, la histoplasmosis, la sarcoidosis, la lesión por los AINEs y las enfermedades infiltrativas del intestino delgado (el linfoma no Hodgkin y la amiloidosis). La mucosa del duodeno con frecuencia tiene vellosidades dilatadas con los vasos linfáticos ectásicos y un puntillado color amarillo pálido. El tratamiento es antibiótico, especialmente para aquellos que atraviesan la barrera hematoencefálica por el frecuente compromiso del SNC. Se recomienda ceftriaxona 2g/día por 2 semanas, seguido de TMP/SMX 160/800 2 veces al día por 1 a 2 años. Una alternativa es la doxiciclina (200 mg/día) más hidroxicloroquina (600 mg/día) vía oral por un año.28, 30
Esprúe tropical
El esprúe tropical es considerado una enfermedad endémica de ciertas partes del mundo. Afecta a los residentes y a los visitantes de áreas, como el sur de Asia, el Caribe, América Central y del Sur. Los pacientes típicamente se presentan con una diarrea profusa, pérdida de peso y malabsorción, que comúnmente resulta en deficiencias de folato y vitamina B12. Se cree que una infección bacteriana, viral o parasitaria es la causa. Bacterias, incluidas en las especies de Klebsiella, Enterobacter y Escherichia, han sido identificadas en los pacientes con esprúe tropical, aunque aún no ha sido identificado un patógeno causante. Como tratamiento se ha propuesto la terapia con antibióticos de amplio espectro y suplementar con ácido fólico y vitamina B12. La biopsia duodenal en el 75% de los casos muestra la AV parcial y rara vez se observa la AV completa. El LIE está típicamente presente, con un recuento medio de aproximadamente el 77%. El número de eosinófilos en la lámina propia es mayor en relación a lo encontrado en la EC (26/HPF vs. 14/HPF). Los cambios histopatológicos en el esprúe tropical son generalmente mucho más prominentes en el ID distal. Los anticuerpos anti-tTG son negativos.31
Giardiasis
Giardia duodenalis es una causa común de enfermedades gastrointestinales. Se caracteriza por generar AV y el síndrome de malabsorción. Frecuentemente causa una diarrea crónica en los pacientes con déficit de IgA o IDCV. Tradicionalmente, el diagnóstico de la infección por Giardia se realiza detectando la presencia de quistes/trofozoítos del parásito en la microscopía de las heces, que se considera el estándar de oro. Sin embargo, la sensibilidad de esta técnica es baja y depende de la carga parasitaria y la experiencia del observador. Se han probado varias técnicas nuevas para mejorar el rendimiento, como ensayos serológicos para la detección de los antígenos utilizando pruebas de fluorescencia directa, ELISA y pruebas inmunocromatográficas, como así también el análisis molecular por PCR de muestras de heces. Las muestras de aspirado duodenal y biopsia duodenal obtenidas endoscópicamente pueden utilizarse para su detección mediante la citología, el examen histológico y las técnicas moleculares. La biopsia duodenal puede ser positiva para Giardia incluso en ausencia del parasito en las heces, ya que puede haber una excreción intermitente del mismo. En el análisis histopatológico se suele detectar al parásito sobre el epitelio en la luz intestinal. La Giardia tiene forma de pera o de hoz, dependiendo del plano de sección, y es un organismo binucleado.31
Esprúe colágeno
El esprúe colágeno (SC) produce la malabsorción y la diarrea. Comparte la mayoría de las características histológicas encontradas en la EC, pero también demuestra característicamente la capa engrosada de colágeno adyacente a la superficie epitelial. El espesor normal de la banda de colágeno en el duodeno es ≤ 5 µ; en el SC, la banda de colágeno es gruesa, irregular, a menudo envuelve los capilares y puede resultar en el desprendimiento del epitelio superficial. Los pacientes con SC refractario pueden requerir un tratamiento con corticosteroides o inmunosupresores. Los cambios histológicos pueden persistir después del tratamiento. Como se señaló anteriormente, el hallazgo de una banda de colágeno debe impulsar a una revisión cuidadosa de la medicación (olmesartán/AINEs).23, 31
Diarrea crónica inflamatoria o exudativa
La diarrea inflamatoria se caracteriza por deposiciones frecuentes, las heces de pequeño volumen con sangre o pasaje de moco y pueden estar acompañadas por síntomas como pujos, tenesmo, fiebre o dolor abdominal severo. Se sospecha una diarrea inflamatoria con la demostración de leucocitos o proteínas leucocitarias (por ejemplo: calprotectina o lactoferrina) en el examen de las heces. Otros estudios de laboratorio que pueden indicar una diarrea inflamatoria incluyen la PCR, la velocidad de sedimentación elevadas y el nivel de albúmina bajo en suero. Las causas más frecuentes son: la EII (enfermedad de Crohn o colitis ulcerosa), la colitis isquémica y los procesos infecciosos, como el C. difficile, el citomegalovirus, la tuberculosis o la Entamoeba histolytica. La colitis por radiación y neoplasias son causas poco frecuentes de la diarrea inflamatoria. El estudio inicial en estos pacientes debe ser la colonoscopia con ileoscopia para la evaluación del íleon terminal (fundamental en sospecha de la EII) y toma de biopsias. La VCC será de utilidad para descartar otros diagnósticos como la colitis isquémica, por radiación, pseudomembranosa, la TBC, la amebiasis y el cáncer colorrectal (CCR).
Causas de diarrea inflamatoria o exudativa:
Enfermedad inflamatoria intestinal:
- colitis ulcerosa/enfermedad de Crohn;
- diverticulitis;
- yeyunoileítis ulcerativa;
Enfermedades infecciosas:
- bacterias: Clostridium difficile;
- virus: herpes simple, CMV;
- parásitos: Amebiasis, strongyloides;
Colitis isquémica.
Colitis por radiación.
Neoplasias:
- CCR/linfoma.
Clostridium difficile (CD)
El CD es una bacteria grampositiva, anaerobia estricta, productora de toxinas y de esporas, principal agente etiológico de la diarrea asociada a los antibióticos. La infección por C. difficile (ICD) se caracteriza por poseer un diferente espectro clínico que va desde los portadores asintomáticos, las formas autolimitadas leves, a formas graves: colitis pseudomembranosa y complicaciones severas como megacolon tóxico, colitis fulminante y sepsis. Los factores de riesgo para la ICD relacionados con el paciente son la exposición a los antibióticos, la edad avanzada (> 65 años) y la hospitalización. Otros grupos de riesgo son las embarazadas, los pacientes inmunosuprimidos, DBT, IRC, el trasplante de órganos sólidos y los pacientes con la EII (colitis ulcerosa). Casi todo antibiótico se ha asociado con el desarrollo de la ICD, incluidos metronidazol y vancomicina. Sin embargo, penicilinas, cefalosporinas, clindamicina y fluoroquinolonas poseen un mayor riesgo de inducción de ICD que otros antibióticos. Tras la ingestión, las esporas resisten el pH bajo del estómago y alcanzan el ambiente anaeróbico del intestino. Cuando el microbioma intestinal se altera debido al tratamiento con los antibióticos, las esporas germinan. A continuación, el CD se desarrolla en su forma vegetativa, prolifera, y coloniza el intestino. La ICD es una enfermedad intestinal mediada por potentes enzimas citotóxicas que dañan la mucosa del intestino. Las cepas patógenas producen múltiples toxinas. Las toxinas principales son la toxina A (enterotoxina) y la toxina B (citotoxina). Ambas son proteínas de alto peso molecular (308 y 270 kDa, respectivamente) que tienen la capacidad de unirse a algunos receptores específicos en las células de la mucosa intestinal del huésped. El daño por la toxina de la mucosa del colon lleva a una acumulación de fibrina, mucina y células muertas; finalmente estructurando una capa de detritus celulares en el colon. Las toxinas también inducen la liberación de citoquinas proinflamatorias por enterocitos, mastocitos y macrófagos. Las características clínicas de la ICD, además de la diarrea acuosa, incluyen el dolor abdominal, la fiebre, las náuseas, los vómitos, la debilidad y la hiporexia. En las formas graves puede presentarse la deshidratación significativa, la distensión abdominal, la hipoalbuminemia con edema periférico y el subsiguiente shock circulatorio. Otras complicaciones graves de la ICD incluyen el megacolon tóxico, la perforación del colon, el íleo, la insuficiencia renal, el síndrome de respuesta inflamatoria sistémica (SIRS), la septicemia y la muerte. La tasa de mortalidad debida a la ICD se estima en el 5%, mientras que la mortalidad asociada con las complicaciones alcanza el 15-25% y hasta el 34% en las unidades de cuidados intensivos (UCI). Una consideración importante en la ICD es la creciente incidencia de la aparición de una cepa hipervirulenta de la bacteria, conocida como ribotipo 027, NAP1. La cepa del CD BI/NAP1/027 es resistente a las fluoroquinolonas, presenta una intensa producción de esporas, aumento de la producción de toxinas A y B y es responsable de los casos más graves de la ICD. La enfermedad moderada a grave por el CD se acompaña generalmente de síntomas sistémicos como el dolor abdominal cólico, la fiebre, la leucocitosis (hasta 50000 células/mm3) y la hipoalbuminemia (< 2,5 mg/dl). Puede observarse la pared del colon engrosada en la tomografía computada (TC) y en la mitad de los casos las pseudomembranas se pueden ver con la endoscopia. Son lesiones elevadas, blancas a amarillas, típicamente de unos 2 cm de diámetro, que se distribuyen irregularmente y separadas por mucosa normal. No se eliminan por lavado de la pared intestinal. No todos los pacientes con la ICD tienen pseudomembranas y su ausencia no descarta la ICD. Por ejemplo, las pseudomembranas rara vez se encuentran en la ICD recurrente o en los pacientes con EII. Por otro lado, una colitis pseudomembranosa puede ser causada por una serie de etiologías diferentes (enfermedad de Behcet, colitis colágena, EII, colitis isquémica, colitis por CMV o Escherichia coli enterohemorrágica O157: H7). El diagnóstico de megacolon tóxico se basa en la evidencia radiológica de la dilatación del colon (> 6 cm), comúnmente involucrando al colon ascendente o transverso. Las imágenes del abdomen (rayos X, ultrasonido) en los pacientes con la ICD revelan asas intestinales distendidas, a menudo con engrosamiento de la pared. La TC de abdomen y pelvis con contraste oral y endovenoso es útil en los pacientes con ICD grave, ya que ayuda a evaluar la presencia de megacolon, perforación intestinal u otros hallazgos, justificando una intervención quirúrgica. Diagnóstico: para interpretar los resultados del laboratorio, deben tenerse en cuenta las técnicas que se aplicaron. El diagnóstico de la ICD está basado principalmente en los signos y síntomas clínicos y sólo confirmado por las pruebas de laboratorio. El cultivo toxigénico, es decir, el cultivo anaeróbico en un medio especial, combinado con la evidencia de la toxina en el sobrenadante de cultivo, se considera como la prueba estándar de oro para el diagnóstico. Esta prueba requiere más de 72 horas para llegar al resultado, por lo que no es un método adecuado en el diagnóstico agudo. El CD produce y secreta la enzima glutamato deshidrogenasa (GDH); esta enzima permite que la bacteria controle el estrés oxidativo derivado de la respuesta inmune, inactivando el peróxido de hidrógeno a través de la producción de α-cetoglutarato. Aunque el cribado de muestras de las heces para GDH en el diagnóstico de ICD es común, su valor se limita a ser una prueba preliminar, ya que tanto las cepas toxigénicas como las no toxigénicas producen GDH. El Test GDH (EIA) (demora en el resultado < 2 h) se considera una prueba de detección inicial con alta sensibilidad y alto VPN. Las muestras positivas para GDH deben ser sometidas a una prueba de confirmación de la infección toxigénica, ya sea por determinación de la toxina A y B (EIA) (demora < 2 h) o por la prueba de amplificación de ácidos nucleicos NAAT (PCR) (Tiempo estimado < 4 h). El ensayo de neutralización de las citotoxinas (CTNA) rara vez se usa para el diagnóstico de rutina, debido a que su resultado demora y a su bajo potencial de estandarización y automatización (Tabla 6). Ninguna prueba es adecuada como prueba independiente para confirmar la ICD. La mejor manera de optimizar el diagnóstico es combinar dos pruebas en un algoritmo. La primera prueba debe ser una prueba con alto VPN (puede ser un GDH, EIA o NAAT). La segunda prueba debe tener un alto VPP (toxina A/B EIA). Si la primera prueba es negativa, excluye la ICD. Si la primera prueba es positiva, debe realizarse la segunda prueba (toxina A/B EIA). Si la segunda prueba es positiva, confirma la ICD. Si la segunda prueba es negativa, el caso necesita ser evaluado clínicamente y el resultado puede deberse a tres situaciones: la ICD con niveles de toxinas por debajo del umbral de detección, un resultado falso negativo para toxina A/B o un portador del CD. El algoritmo sugerido del diagnóstico de la ICD se presenta en la Figura 5. Tratamiento: las guías recomiendan el uso de metronidazol para tratar un episodio inicial de la ICD leve a moderado. La dosis es de 500 mg por vía oral, 3 veces al día durante 10 a 14 días. En un caso grave, vancomicina (125 mg por vía oral, 4 veces al día durante 10 a 14 días). Una combinación de vancomicina oral (500 mg 4 veces al día) y metronidazol intravenoso (500 mg cada 8 h) están indicados para el tratamiento de la ICD grave y complicada. Por otro lado, la ACG recomienda una dosis de 125 mg de vancomicina.32 Al tratar un primer episodio de recurrencia, el régimen debe ser el mismo que un caso inicial, según la gravedad de la infección. Si hay un segundo episodio de recurrencia, se recomienda un pulso de vancomicina. La interpretación de los datos del laboratorio es complicada, ya que la presencia del C. difficile en las heces no significa la ICD; y al revés, la ausencia de toxinas no descarta la posibilidad de la ICD. La detección de la toxina no es indicativa de un peor pronóstico. La noción de que un ensayo de toxina puede distinguir entre colonización e infección es falsa; esta diferenciación es un desafío clínico y no puede basarse únicamente en la evaluación del laboratorio. El riesgo de recurrencia de la ICD después del tratamiento inicial es aproximadamente del 20 al 25%. El trasplante de microbiota fecal (FMT) se está estudiando como una alternativa a la terapia con los antibióticos estándar para tratar la ICD recurrente. Las opciones potenciales adicionales para el tratamiento de la ICD incluyen rifaximina, nitazoxanida, colestiramina, inmunoglobulina intravenosa (IGIV), anticuerpos monoclonales, vacunas y probióticos. Los diversos probióticos comúnmente utilizados incluyen especies de Lactobacilos, Bifidobacterium y Saccharomyces. Actualmente, los probióticos no tienen ningún papel en la prevención primaria de la ICD. Saccharomyces boulardii produce una enzima que inhibe los efectos de las toxinas A y B de C. difficile.
Tabla 6. Pruebas diagnósticas para la infección por Clostridium difficile
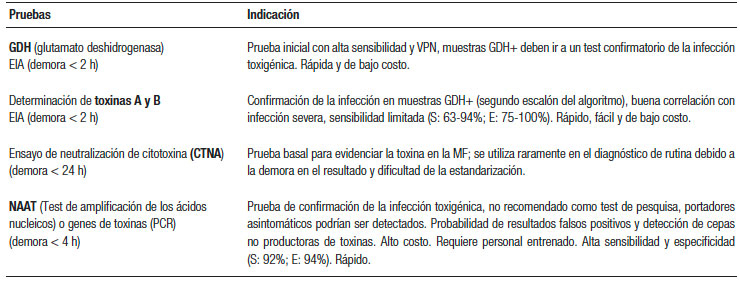
Allegretti et al. mostraron una mayor concentración de AB secundarios en las heces de personas sanas comparada con la ICD, mientras que la concentración de AB primarios fue mayor en los pacientes con la ICD recurrente en comparación con los pacientes con su primer episodio de infección. La influencia de los AB es probablemente más compleja que el modelo simple donde los AB primarios estrictamente promueven y los secundarios inhiben la germinación y la vegetación del CD como se describe en la excelente revisión de Baktash et al.33-36
Figura 5. Algoritmo diagnóstico ante la sospecha de diarrea por Clostridium spp.
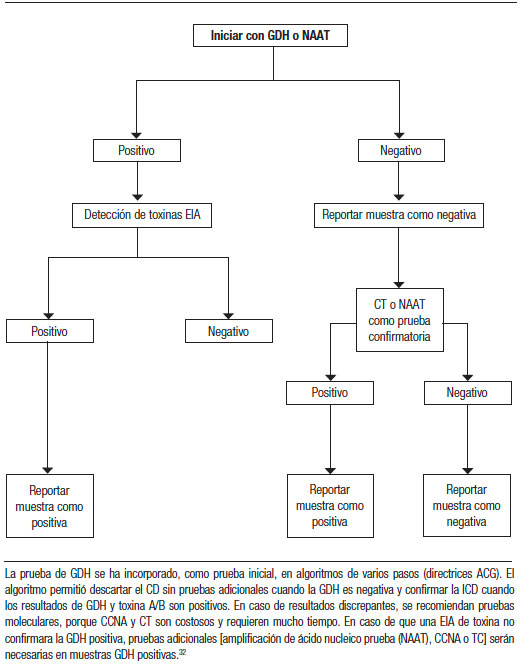
Causas de diarrea en los pacientes con diabetes mellitus (DM)
Las complicaciones gastrointestinales de la DBT incluyen: gastroparesia, constipación (60%), diarrea (20%), incontinencia fecal e hígado graso no alcohólico. En los pacientes con DBT II de larga data la diarrea crónica puede presentarse como consecuencia de diferentes mecanismos; ellos son: secundario a drogas utilizadas en el tratamiento de la DBT (metformina, estatinas, repaglinida, rosigilitazona, meglitinidas, agonistas del receptor GLP-1 en el 20%, inhibidores de la DDP-4 hasta en el 10%, etc.), la EC asociada, la IPE, el SBID por dismotilidad (40%), la incontinencia por afectación del esfínter, disfunción anorrectal (neuropatía DBT), la MAB y la malabsorción de hidratos de carbono (alimentos dietéticos ricos en fructosa o sorbitol). La hiperglucemia y la variación de la glucemia puede influir en la función del esfínter. La hiperglucemia aguda inhibe la función del esfínter anal y disminuye la compliance del recto llevando a la incontinencia fecal. Es importante conocer la fisiopatología de la gastroenteropatía diabética que es multifactorial. Además de la neuropatía autonómica que causa las alteraciones gastrointestinales, el papel del sistema nervioso entérico (SNE) es cada vez más evidente. Se han estudiado diferentes mecanismos potenciales, comprometiendo al SNE en los trastornos de la motilidad gastrointestinal en la DBT. El SNE está estructurado en dos plexos principales, mientérico y submucoso, formados por pequeños ganglios y neuronas conectadas a través de haces de nervios que recorren todo el tracto GI. Este sistema está conectado al sistema nervioso central a través de neuronas sensoriales que envían fibras aferentes que transmiten sensaciones viscerales como el dolor, el estiramiento, la plenitud y las náuseas, así como vías eferentes simpáticas y parasimpáticas que modulan la motilidad, la secreción y la circulación. Además de las neuronas entéricas, las células intersticiales de Cajal (ICC) son células no neuronales y no gliales que están presentes en todo el tracto GI dentro de múltiples capas de la pared esofágica, gástrica e intestinal, incluido el plexo mientérico. Estas células funcionan como “marcapasos”, generando actividad eléctrica que resulta en el movimiento peristáltico de onda lenta del intestino. Además, están involucrados en la neurotransmisión entre las neuronas motoras entéricas, las eferentes del sistema nervioso central y las células del músculo liso en la pared del tracto GI. La neurotransmisión inhibitoria en el SNE ocurre a través de la vía no adrenérgica no colinérgica (NANC). El óxido nítrico (ON) es el principal neurotransmisor inhibidor. Los estudios de los modelos animales con DM han demostrado que las causas de los trastornos gastrointestinales motores se deben no solo a la neuropatía autonómica, sino también al incremento del estrés oxidativo y de la apoptosis, pérdida de factores de crecimiento y reducción de células intersticiales de Cajal, cambios en la microbiota que pueden modular el eje cerebro intestinal y la disfunción endotelial en la microvasculatura. En el SNE habría un desbalance entre las neuronas inhibitorias y las excitatorias, acumulación de especies reactivas de oxígeno, que sinérgicamente causan estrés oxidativo y llevan a la apoptosis. Los fenómenos de remodelación posteriores (los cambios estructurales en la matriz del tejido conectivo y músculo liso intestinal, atrofia y degeneración de fibras nerviosas) llevan a los trastornos en la motilidad y la secreción. La estrategia terapéutica en estos pacientes consiste en 1) la adecuada hidratación e inicio de agentes antidiarreicos como loperamida, 2) mejorar el control glucémico (consulta al nutricionista y el uso de alternativas como terapia con bomba de insulina y monitoreo continuo de glucemia), 3) si los síntomas persisten TAE para SBID o prueba terapéutica con rifaximina) y 4) considerar la IPE (prueba de elastasa fecal). Orientan al diagnóstico de neuropatía autonómica síntomas como: la respuesta pupilar anormal, la anhidrosis, la salivación, la hipotensión ortostática, la impotencia, la eyaculación retrograda, la disfunción vesical urinaria y las parestesias en pies y piernas.37-39
Diarrea en el anciano
En esta población, se deben tener en cuenta en el diagnóstico diferencial de las causas de diarrea crónica los siguientes: la colitis microscópica (lansoprazol, AAS, sertralina), la impactación fecal, el CCR, la colitis isquémica, la diverticulosis y el SBID (aclorhidria y gastritis atrófica). Será fundamental la realización del tacto rectal y el examen proctológico. Estar especialmente atentos a la presencia de dolor abdominal y la evaluación de los pulsos periféricos ante sospecha de patología vascular. Los estudios endoscópicos (VCC) son necesarios en este grupo etario. Habitualmente existen comorbilidades que favorecen el desarrollo de la diarrea como la DBT, la enfermedad de Parkinson, el hipotiroidismo, entre otras. Es fundamental, además, tener en cuenta los medicamentos habituales como posibles causas de diarrea.
En conclusión, el estudio de los pacientes con diarrea crónica muchas veces nos enfrenta a un verdadero desafío, suele ser complejo y las causas multifactoriales. Sin embargo, el conocimiento de las herramientas diagnósticas y terapéuticas disponibles nos ayuda a realizar un enfoque adecuado, orientado a las posibles causas, costo-efectivo y razonable. Muchas veces estaremos tratando los mecanismos fisiopatológicos de la diarrea, mientras avanza el proceso diagnóstico hasta llegar a la enfermedad de origen.
Figura 6. Algoritmo diagnóstico para el enfoque clínico inicial del estudio de la diarrea crónica
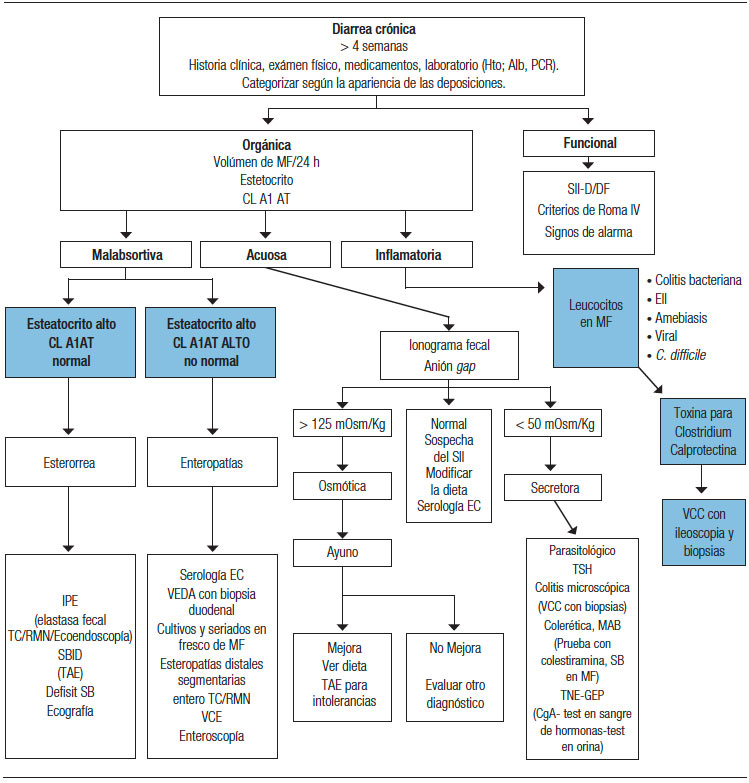
Referencias
- Schiller LR, Pardi DS, Sellin JH. Chronic Diarrhea: Diagnosis and Management. Clinical Gastroenterology and Hepatology 2017; 15 (2): 182-193.e3. Disponible en: http://dx.doi.org/10.1016/j.cgh.2016.07.028
- Arasaradnam RP, Brown S, Forbes A, Fox MR, Hungin P, Kelman L, et al. Guidelines for the investigation of chronic diarrhoea in adults: British Society of Gastroenterology, 3rd edition. Gut 2018; 67 (8): 1380-1399.
- Ong J, Swift C, Norman R, Allwood I, Al-Naeeb Y, Shankar A. The investigation of chronic diarrhoea: new BSG guidance. Br J Gen Pract 2019; 69 (682): 262-264. Disponible en: https://doi.org/10.3399/bjgp19X702653
- Menees S, Chey W. The gut microbiome and irritable bowel syndrome. F1000Res 2018; 7: 1029. Disponible en: http://dx.doi.org/10.12688/f1000research.14592.1
- Di Costanzo M, Berni Canani R. Lactose Intolerance: Common Misunderstandings. Ann Nutr Metab 2018; 73 (4): 30-37. Doi: 10.1159/000493669.
- Ianiro G, Cammarota G, Valerio L, Annicchiarico BE, Milani A, Siciliano M, Gasbarrini A. Microscopic colitis. World J Gastroenterol 2012; 18 (43): 6206-6215. Doi: 10.3748/wjg.v18.i43.6206
- Villanacci V, Mourin AJ, Salviato T. Microscopic Colitis. En: Jouret-Mourin A, Faa G, Geboes K (eds). Nueva York: Springer 2018: 199-213. Disponible en: http://dx.doi.org/10.1007/978-3-319-89503-1_14
- Nguyen GC, Smalley WE, Vege SS, Carrasco-Labra A, Clinical Guidelines Committee. American Gastroenterological Association Institute Guideline on the Medical Management of Microscopic Colitis. Gastroenterology 2016; 150 (1): 242-246. Disponible en: http://dx.doi.org/10.1053/j.gastro.2015.11.008
- Pardi DS, Tremaine WJ, Carrasco-Labra A. American Gastroenterological Association Institute Technical Review on the Medical Management of Microscopic Colitis. Gastroenterology 2016: 150: 247-274. Disponible en: http://dx.doi.org/10.1053/j.gastro.2015.11.006
- O’Connor J, Acosta Haab G, Armas I, Belli S, et al. Recomendaciones para el diagnóstico y el tratamiento de tumores neuroendocrinos de origen gastroenteropancreáticos. Acta Gastroenterol Latin 2020; 50 (1): 1-33.
- Basso S, Canicoba M, Capitanich P, Carballido M, et al. I Consenso argentino de diagnóstico y tratamiento de la insuficiencia pancreática exocrina. Acta Gastroenterol latinoam 2018; 48 (3): 213-225.
- Rezaie A, Buresi M, Lembo A, Lin H, et al. Hydrogen and Methane-Based Breath Testing in Gastrointestinal Disorders: The North American Consensus. Am J Gastroenterol 2017; 112 (5): 775-784. Doi: 10.1038/ajg.2017.46
- Camilleri M. Bile Acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liver 2015; 9 (3): 332-339. Disponible en: http://dx.doi.org/10.5009/gnl14397
- Oduyebo I, Camilleri M. Bile acid disease: the emerging epidemic. Current Opinion in Gastroenterology 2017; 33 (3): 189-195. Disponible en: http://dx.doi.org/10.1097/mog.0000000000000344
- Camilleri M, Sellin JH, Barrett KE. Pathophysiology, Evaluation, and Management of Chronic Watery Diarrhea. Gastroenterology 2017; 152 (3): 515-532. Doi: 10.1053/j.gastro.2016.10.014
- Walters JR, Pattni SS. Managing bile acid diarrhoea. The-rap Adv Gastroenterol 2010; 3 (6): 349-357. Doi: 10.1177/1756283X10377126
- Fernández-Bañares F, Esteve M, Salas A, Alsina M, Farré C, González C, Buxeda M, Forné M, Rosinach M, Espinós JC, Maria Viver J. Systematic evaluation of the causes of chronic watery diarrhea with functional characteristics. Am J Gastroenterol 2007; 102 (11): 2520-2528. Doi: 10.1111/j.1572-0241.2007.01438.x
- Barkun AN, Love J, Gould M, Pluta H, Steinhart H. Bile acid malabsorption in chronic diarrhea: pathophysiology and treatment. Can J Gastroenterol 2013; 27 (11): 653-659.
- Murray JA, Rubio-Tapia A. Diarrhea due to small bowel diseases. Best Pract Res Clin Gastroenterol 2012; 26 (5): 581-600. Doi: 10.1016/j.bpg.2012.11.013
- Deane S, Selmi C, Naguwa SM, Teuber SS, Gershwin ME. Common variable immunodeficiency: etiological and treatment issues. Int Arch Allergy Immunol 2009; 150 (4): 311-324. Doi: 10.1159/000226232
- Agarwal S, Smereka P, Harpaz N, Cunningham-Rundles C, Mayer L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm Bowel Dis 2011; 17 (1): 251-259. Doi:10.1002/ibd.21376
- Rubio-Tapia A, Herman ML, Ludvigsson JF, Kelly DG, Mangan TF, Wu TT, Murray JA. Severe spruelike enteropathy associated with olmesartan. Mayo Clin Proc 2012; 87 (8): 732-738. Doi: 10.1016/j.mayocp.2012.06.003
- Kamboj AK, Oxentenko AS. Clinical and Histologic Mimickers of Celiac Disease. Clin Transl Gastroenterol 2017; 8 (8): e114. Doi:10.1038/ctg.2017.41
- Ianiro G, Bibbò S, Montalto M, Ricci R, Gasbarrini A, Cammarota G. Systematic review: Sprue-like enteropathy associated with olmesartan. Aliment Pharmacol Ther 2014; 40 (1): 16-23.
- Ianiro G, Gasbarrini A, Cammarota G. Olmesartan-associated sprue-like enteropathy: know your enemy. Scand J Gastroenterol 2016; 51 (7): 891.
- Ridolo E, et al. Eosinophilic disorders of the gastro-intestinal tract: an update. Clin Mol Allergy 2016; 14: 17. Doi 10.1186/s12948-016-0055-y
- Abou Rached A, El Hajj W. Eosinophilic gastroenteritis: Approach to diagnosis and management. World J Gastrointest Pharmacol Ther 2016; 7 (4): 513-523. Doi: 10.4292/wjgpt.v7.i4.513
- Dolmans RAV, Boel CHE, Lacle MM, Kusters JG. Clinical Manifestations, Treatment, and Diagnosis of Tropheryma whipplei Clin Microbiol Rev 2017; 30 (2): 529-555. Disponible en: https://doi.org/10.1128/CMR.00033-16
- Moos V, Loddenkemper C, Schneider T. Infektionen mit Tropheryma whipplei. Besiedlung, selbstlimitierte Infektion und Morbus Whipple [Tropheryma whipplei Colonization, self-limiting infection and Whipple’s disease]. Pathologe 2011; 32 (5): 362-670. German. Doi: 10.1007/s00292-011-1446-y
- Bureš J, Kopáčová M, Douda T, Bártová J, Tomš J, Rejchrt S, Tachecí I. Whipple’s Disease: Our Own Experience and Review of the Literature. Gastroenterol Res Pract 2013; 2013: 478349. Doi: 10.1155/2013/478349
- Owen DR, Owen DA. Celiac Disease and Other Causes of Duodenitis. Arch Pathol Lab Med 2018; 142 (1): 35-43. Doi: 10.5858/arpa.2016-0608-RA
- Surawicz CM, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile Am J Gastroenterol 2013; 108 (4): 478-498.
- Martínez-Meléndez A, Camacho-Ortiz A, Morfin-Otero R, Maldonado-Garza HJ, Villarreal-Treviño L, Garza-González E. Current knowledge on the laboratory diagnosis of Clostridium difficile World J Gastroenterol 2017; 23 (9):1552-1567. Disponible en: http://dx.doi.org/10.3748/wjg.v23.i9.1552
- Chambers J, Gibbs T, Speers D, Riley T. The laboratory diagnosis of Clostridium difficile Pathology 2019; 51: S57. Disponible en: http://dx.doi.org/10.1016/j.pathol.2018.12.128
- Aljarallah KM. Conventional and alternative treatment approaches for Clostridium difficile Int J Health Sci 2017; 11 (1): 1-10.
- Czepiel J, Dróżdż M, Pituch H, Kuijper EJ, Perucki W, Mielimonka A, et al. Clostridium difficile infection: review. Eur J Clin Microbiol Infect Dis 2019; 38 (7): 1211-1221.
- Maisey A. A Practical Approach to Gastrointestinal Complications of Diabetes. Diabetes Ther 2016; 7: 379-386. Doi: 10.1007/s13300-016-0182-y
- Yarandi S, Srinivasan SS. Diabetic gastrointestinal motility disorders and the role of enteric nervous system: current status and future directions. Neurogastroenterol Motil 2014; 26 (5): 611-624. Doi:10.1111/nmo.12330
- Chandrasekharan B, Srinivasan S. Diabetes and the enteric nervous system. Neurogastroenterol Motil 2007; 19 (12): 951-960. Doi:10.1111/j.1365-2982.2007.01023.x
Correspondencia: María Laura Moreno
Correo electrónico: eballerga@gmail.com
Acta Gastroenterol Latinoam 2020;50(4):388-414
