Rubén Muñoz Cedeño,3 Michelle Ricaurte Enriquez,4 Priscila Martínez Ballesteros,4 Viviana Paullán Sani,1 Gema Rodríguez Chica4
1 Gastroenterólogo del Hospital de Especialidades Dr. Abel Gilbert Pontón, Universidad de Guayaquil, Ecuador.
2 Posgradista de Gastroenterología del Hospital de Especialidades Dr. Abel Gilbert Pontón, Universidad de Guayaquil, Ecuador.
3 Médico General del Hospital de Especialidades Dr. Abel Gilbert Pontón, Universidad Católica de Santiago de Guayaquil, Ecuador.
4 Médico General del Hospital Universitario de Guayaquil de la Universidad de Guayaquil, Ecuador.
Acta Gastroenterol Latinoam 2020;50(4):479-484
Recibido: 27/07/2019 / Aprobado: 18/09/2020 / Publicado en www.actagastro.org el 14/12/2020

Resumen
El síndrome de poliposis juvenil (SPJ) es una enfermedad autosómica dominante caracterizada por múltiples pólipos juveniles en el tracto gastrointestinal. Estos pólipos pueden estar presentes en todo el tracto gastrointestinal, generalmente en el colon. Presentamos el caso de un adolescente de 16 años de edad con antecedentes personales de pólipos y de una cirugía para resección de intestino por cuadro de abdomen obstructivo en dos ocasiones (a los 5 y 11 años de edad). Se programan estudios endoscópicos donde se encuentran pólipos y se realiza polipectomía. A las 24 horas presentó proctorragia y cuadro de abdomen agudo obstructivo que fue intervenido por laparotomía exploratoria que mostró intususcepción.
Palabras claves. Poliposis juvenil, polipectomía endoscópica, abdomen agudo, pólipos.
Acute Abdomen in a Teenager with Juvenile Polyposis. About a Case
Summary
Juvenile polyposis syndrome is an autosomal-dominant condition disease characterized by multiple juvenile polyps in the gastrointestinal tract. These polyps may be present in the entire digestive tract; generally in the colon. We present the case of a 16-year-old adolescent with a history of polyps, who had had bowel resections for acute abdomen on two occasions (at 5 and 11 years of age). Endoscopic procedures were carried out, finding polyps and performing a polypectomy. After 24 hours, the patient presented proctorrhagia associated with acute abdominal pain by an obstruction, for which he was surgically intervened with an exploratory laparotomy where an intussusception was found.
Key words. Juvenile Polyposis, endoscopic polypectomy, acute abdomen, polyps.
Introducción
El síndrome de poliposis juvenil (SPJ) es una enfermedad autosómica dominante del tracto gastrointestinal, caracterizada por múltiples pólipos juveniles asociados al desarrollo de cáncer gastrointestinal en un 10 a 50% de los casos. El más común es el cáncer colorrectal; aparece antes de los 20 años de edad y su incidencia es de 1 por cada 100.000-160.000 habitantes/año a nivel mundial.1
La clínica de la enfermedad varía, presentando hemorragia gastrointestinal, anemia, dolor abdominal, rectorragia y diarrea. Aproximadamente un 15% de los pacientes presenta manifestaciones extraintestinales, como mal rotación intestinal, alteraciones cardíacas (por ejemplo prolapso de la válvula mitral) o cerebrales (macrocefalia, hidrocefalia), paladar hendido, polidactilia y/o alteraciones en genitales o en el tracto urinario.1 Los criterios diagnósticos son: presencia de cinco o más pólipos juveniles en el colon o recto, o pólipos juveniles en otros segmentos del tracto gastrointestinal, o cualquier número de pólipos juveniles con un antecedente familiar.2
La colonoscopía es el método diagnóstico de elección, ya que además de identificar las lesiones permite su resección. Los pólipos resecados deben ser enviados a anatomía patológica, para identificar su histología y descartar sospecha de malignidad o enfermedad sindrómica.3 En caso de displasia intestinal o de hallarse un adenocarcinoma, es recomendable la colectomía total o subtotal, según el grado de afectación.4 Aproximadamente el 33% de los pacientes tienen un progenitor con la enfermedad.5
La cirugía profiláctica es considerada en pacientes que presentan: poliposis colorrectal no tratable por endoscopía (más de 50-100 pólipos), síntomas severos de hemorragia gastrointestinal o diarrea; pólipos juveniles displásicos, y pacientes con historia familiar de cáncer colorrectal. Las cirugías propuestas son la colectomía subtotal con anastomosis ileorrectal o la proctocolectomía total con reservorio.6
Presentación del caso
Se trata de un paciente masculino de 16 años de edad con antecedentes patológicos personales de pólipos y antecedente quirúrgico de resección de intestino delgado en el hospital pediátrico de Guayaquil por cuadro de abdomen obstructivo en dos ocasiones (a los 5 y 11 años de edad). El paciente no refiere antecedentes familiares de enfermedad del tracto digestivo. Se recaba en su historial clínico: videocolonoscopía tres años atrás, con diagnóstico de pólipos colónicos con histología de un pólipo de sigma que reporta pseudopólipo inflamatorio. Acude referido del hospital pediátrico a la consulta externa en nuestra casa de salud, con diagnóstico de pólipos colónicos con anatomía patológica no concluyente. De manera ambulatoria se realiza videoendoscopía digestiva alta, y se observa en duodeno un pólipo lobulado de más de 20 mm de diámetro con tallo largo, no resecado, y tres pólipos gastroduodenales menores de 10 mm, pediculados, que se extraen mediante polipectomía. En la videocolonoscopía se observa en la unión cecoascendente un pólipo pediculado de aspecto arboriforme con tallo pequeño de gran tamaño, que obstruye el 90% de la luz intestinal y no permite continuar la exploración. Se realiza polipectomía de tres pólipos, sin complicaciones, y es dado de alta a la espera de los resultados de la biopsia. A las 24 horas aproximadamente, ingresa a nuestro hospital por cuadro caracterizado por dolor abdominal en marco colónico de moderada a gran intensidad, acompañado de hematemesis de poca cantidad en cinco ocasiones, astenia, presión arterial de 100/60 mmHg, taquicárdico con frecuencia cardíaca de 133 latidos por minuto, afebril. Al examen físico, muestra palidez generalizada, manchas hiperpigmentadas en cavidad bucal y en palmas de manos, cicatrices abdominales en flanco y fosa ilíaca izquierda. En exámenes de laboratorio de ingreso, se reporta anemia con hemoglobina de 4,8 mg/dL y hematocrito de 14,8%, leve leucocitosis de 10400/mm3, con neutrófilos de 68,2%. Se transfunden dos unidades de sangre, con control postransfusional que reporta: leucocitos de
15100/mm3, neutrófilos 79,7%, hemoglobina de
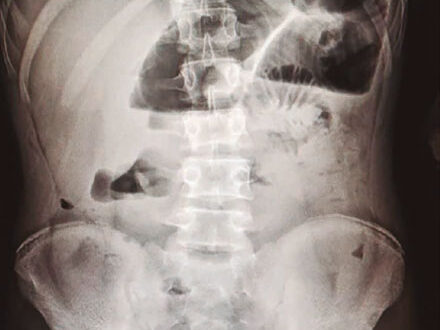
7,5 mg/dL, hematocrito de 22,9%, plaquetas 222.000. En cuanto a la química sanguínea, se reporta: PCR 2,9 mg/dL, electrolitos y función renal normal. Se realiza radiografía abdominal en la que se evidencian niveles hidroaéreos sugestivos de obstrucción intestinal, además de asa centinela (Figura 1).
Figura 1. Radiografía de abdomen simple de pie en la que se evidencian niveles hidroaéreos sugestivos de obstrucción intestinal, además de asa centinela
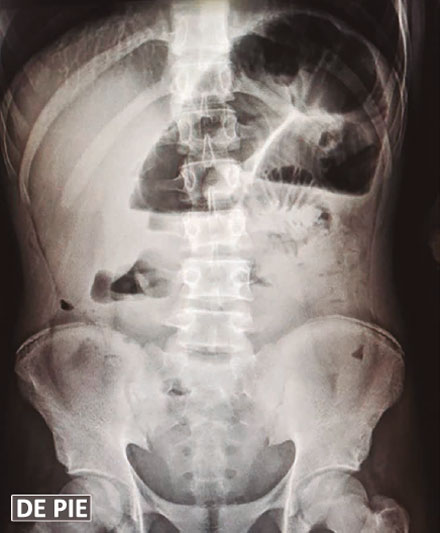
Una ecografía abdominal revela la presencia de asas intestinales distendidas, rellenas de líquido y con reducción del peristaltismo, líquido libre en cavidad, íleo y adherencias peritoneales (Figuras 2 y 3). Además, presenta pico febril de 38,5 ºC. Debido al cuadro, es valorado por el servicio de cirugía general, que define un cuadro de abdomen agudo, por lo que se realiza una laparotomía exploratoria de emergencia con hallazgo de intususcepción (Figura 4). Se realiza resección intestinal con una entero-entero anastomosis latero-lateral de intestino delgado. En el procedimiento se evidencian vestigios de tres anastomosis intestinales antiguas: a 60 y 70 centímetros del ángulo de Treitz y a 140 centímetros de la válvula ileocecal. Se inicia tratamiento antibiótico con piperacilina/tazobactam, con buena respuesta, y descenso progresivo de leucocitos. Cursa postoperatorio con reposición de potasio y calcio por los niveles bajos de los mismos en química sanguínea, con nutrición parenteral hasta el quinto día de postoperatorio. El paciente inicia tolerancia oral con buena respuesta. Al octavo día de postoperatorio, presenta hematemesis de 200 ml, con tres episodios de melena, por lo que se realiza videoendoscopía digestiva alta, y se observan pólipos gástricos sin evidencia de sangrado activo, por lo que es dado de alta, con buena respuesta al tratamiento. La evolución del paciente en los seis meses de seguimiento por la consulta ha sido favorable, sin complicaciones.
Figuras 2 y 3. Ecografía abdominal revela presencia de asas intestinales distendidas, rellenas de líquido y con reducción del peristaltismo observado en la ecografía dinámica e informada en el reporte ecográfico, líquido libre en cavidad, íleo y adherencias peritoneales

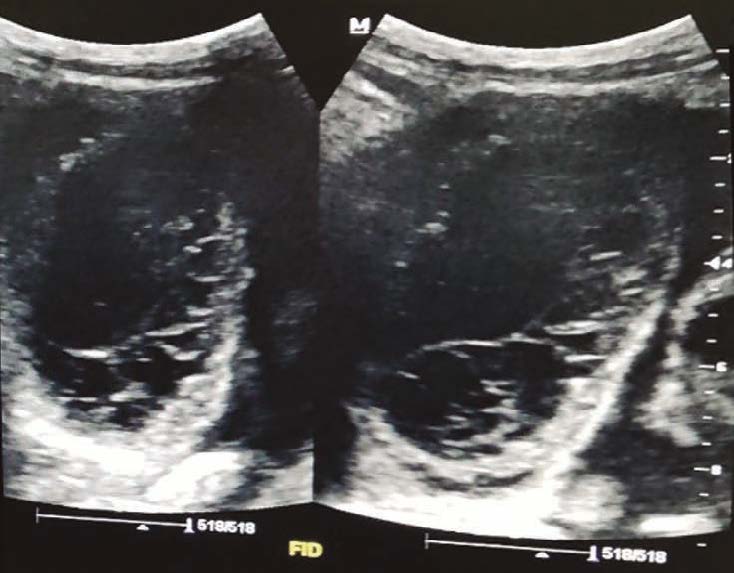
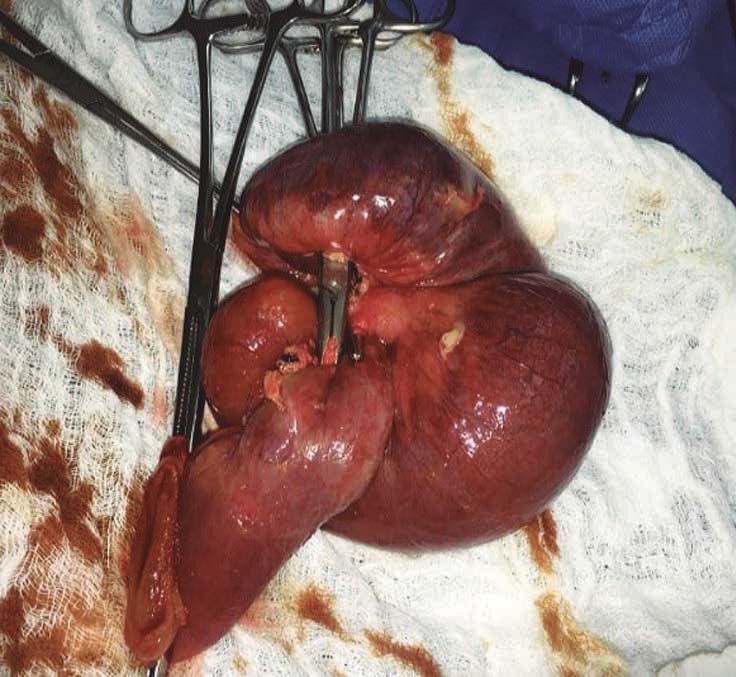
Figura 4. Imagen de aspecto macroscópico; se observan áreas cianóticas del intestino delgado del íleon por abdomen agudo obstructivo por intususcepción, informada por el cirujano en la historia clínica
Discusión
El síndrome de poliposis juvenil (SPJ) es una enfermedad autosómica dominante, considerada una condición premaligna. Se caracteriza por la presentación de múltiples pólipos benignos, que generalmente aparecen antes de los veinte años de edad. Su incidencia es de 1 por cada 100.000 – 160.000 habitantes a nivel mundial.1, 2 Por el tipo de pólipo que se presenta, pertenece a las poliposis hamartomatosas.3
El primer caso de SPJ fue publicado en 1939; se trató de una paciente de treinta meses de edad con pólipos rectales histológicamente estudiados. El término “pólipo juvenil” fue originalmente utilizado en 1957 por Horrilleno y col. En 1962, Morson describió la histología hamartomatosa de los pólipos, diferenciándolos así de los adenomatosos. La condición en general fue considerada benigna hasta que se reportó la malignización de pólipos en 1984.7
Se clasifica clínicamente sobre la base del número de pólipos y su distribución. Los criterios diagnósticos son: presencia de cinco o más pólipos juveniles en el colon o recto, o pólipos juveniles en otros segmentos del tracto gastrointestinal, o cualquier número de pólipos juveniles con un antecedente familiar.2 La edad promedio de diagnóstico es 18,5 años; sin embargo, en muchos casos se diagnostica después.8 Se agrupa en tres presentaciones clínicas que son: la poliposis juvenil en la infancia, la poliposis juvenil colónica y la poliposis juvenil generalizada.4
La poliposis juvenil en la infancia se caracteriza por la presencia de pólipos con distribución difusa en pacientes menores a los seis meses de edad. Por otro lado, la poliposis juvenil colónica se caracteriza por la presencia de múltiples pólipos juveniles localizados en el colon distal y recto, y se presenta en pacientes de 5 a 15 años de edad. Por último, en la poliposis juvenil generalizada se presentan múltiples pólipos en el tracto gastrointestinal, principalmente en el estómago, colon distal y recto. Se da en pacientes de seis meses de edad hasta los 5 años.9
Puede diagnosticarse genéticamente, ya que hasta un 60% de los pacientes posee mutaciones específicas para la enfermedad. Se ha concluido que existen dos mutaciones genéticas relacionadas con la enfermedad: SMAD4 localizado en el cromosoma 18q21 y BMPR1A.10 Ambos genes están involucrados en la vía de señalización del factor de crecimiento transformante beta (TGFB). Ambos sintetizan proteínas y son responsables de procesos que determinan el desarrollo apropiado de la mucosa del colon y, posteriormente, del mantenimiento de la homeostasis en dichos tejidos. La mayoría de mutaciones en estos genes, cuando están presentes en pacientes con SPJ, son deleciones.11
Existe además un síndrome combinado variante fenotípico: la poliposis juvenil/síndrome de telangiectasia hemorrágica (JPHT: Juvenile Polyposis Hereditary Telangiectasia), cuya mutación se encuentra en el gen SMAD4. Dicha variante se presenta en la mayoría de los pacientes con mutaciones identificadas en la línea germinal de los genes BMPR1A y SMAD4. Tiene el mismo riesgo de desarrollar cáncer colorrectal.12, 13
En un estudio por Aytac et al. publicado en el 2015, con un estudio de 35 pacientes con SPJ, se demostró que la mutación SMAD4 está relacionada con pólipos en mayor cantidad (más de cincuenta por paciente), además de comprobarse que está asociada a un mayor riesgo de desarrollo de cáncer digestivo superior más agresivo.14
Debido a que existe un 30-40% de pacientes que no poseen las mutaciones genéticas ya descritas, existen otros genes que han sido investigados y podrían influir en la patogénesis de la enfermedad. Sin embargo, no se han encontrado relaciones germinales con estos otros genes. Por otro lado, existe la mutación en el PTEN que se ha relacionado con el síndrome de Cowden, enfermedad caracterizada por la presencia de múltiples hamartomas en diversos tejidos. Debido a esta manifestación clínica se la considera un diagnóstico diferencial del SPJ y dicho gen ayuda a definir el diagnóstico.6
La clínica de la enfermedad varía; puede presentar hemorragia gastrointestinal, anemia, dolor abdominal, rectorragia y diarrea. Aproximadamente un 15% de los pacientes presenta manifestaciones extraintestinales, como mal rotación intestinal, alteraciones cardíacas (por ejemplo, prolapso de la válvula mitral) o cerebrales (macrocefalia, hidrocefalia), paladar hendido, polidactilia y/o alteraciones en los genitales o en el tracto urinario.1, 2
En un estudio por Latchford et al., del año 2012, se estudió a 44 pacientes, de los cuales: 15 fueron diagnosticados genéticamente, mientras que 29 tuvieron un diagnóstico basándose en su cuadro clínico; 19 pacientes poseían la mutación en el gen SMAD4 y 9 la mutación BMPR1A; 6 pacientes desarrollaron cáncer, 4 pacientes ya tenían cáncer al momento del diagnóstico de SPJ y 3 pacientes desarrollaron cáncer en el tiempo de seguimiento del estudio.15
Un paciente con SPJ puede presentar desde 50 a 200 pólipos, distribuidos principalmente en el colon. Se ha demostrado que la mayoría de los pólipos colónicos están ubicados en el colon distal.8 También pueden encontrarse en el intestino delgado y en el estómago. Su tamaño varía desde milímetros hasta varios centímetros.2 Los pólipos en el SPJ son de tipo hamartomatoso, histológicamente diferentes de los presentes en el síndrome de Peutz-Jeghers. Los pólipos en el SPJ no poseen fibras musculares, como se encuentran histológicamente en los pólipos del síndrome de Peutz-Jeghers.16
Los pólipos juveniles, como se los denomina, no se refieren a la edad de presentación o diagnóstico sino a la histopatología de los mismos. Poseen epitelio de características normales, con un infiltrado inflamatorio de glándulas quísticas de contenido mucoso en túbulos epiteliales de la lámina propia. Este infiltrado también posee fibroblastos y macrófagos. Su apariencia se caracteriza por ser esféricos y lisos, con revestimiento rojizo.2, 10 A los pólipos juveniles típicos del SPJ también se los conoce como pólipos de retención debido al contenido mucoso que presentan.17 Otras características histológicas incluyen la presencia de linfocitos, eosinófilos y neutrófilos en la lámina propia. También se mencionan pólipos atípicos con forma filiforme y multilobulada, con un aumento de la relación glándula-estroma en su estructura.18
El riesgo de malignización es alto en pacientes con SPJ. Cuando existe un pólipo solitario dicho riesgo disminuye y los pacientes no requieren un seguimiento estricto. En un estudio realizado por Howe et al. en 1998, 16 de 29 pacientes (55%) desarrollaron cáncer. De estos, en once se trató de cáncer de colon, en cuatro cáncer de estómago, en uno cáncer de páncreas y otro en la ampolla duodenal.19 En pacientes menores a 18 años, la aparición de cáncer es rara, por lo que no se requiere vigilancia previo a esta edad.2
La colonoscopía es el método diagnóstico de elección, ya que además de identificar las lesiones permite su resección. Los pólipos resecados deben ser enviados a anatomía patológica, para su estudio histopatológico, identificar su histología y descartar sospecha de malignidad o enfermedad sindrómica.3 En un estudio por Brosens et al., del año 2007, se estudió el riesgo de cáncer colorrectal en pacientes con SPJ en comparación con la población general. Los datos recolectados procedían del registro de poliposis en Johns Hopkins. Los pacientes con SPJ tuvieron un riesgo relativo de 34,0 de desarrollar cáncer colorrectal frente a la población general. También se calculó el riesgo de desarrollo de dicho cáncer en pacientes con SPJ, siendo este de 39%.20
En caso de displasia intestinal o de hallarse un adenocarcinoma, es recomendable la colectomía total o subtotal, según el grado de afectación.4 Aproximadamente el 33% de los pacientes tienen un progenitor con la enfermedad,5 por lo que se recomienda el estudio genético de familiares directamente relacionados con el paciente para diagnosticar o descartar la enfermedad.
La colonoscopía con polipectomía se recomienda para prevenir el riesgo de cáncer, sangrado intestinal y obstrucción intestinal. También mejora la anemia del paciente. La vigilancia de los pacientes de riesgo se basa en los signos y síntomas de alarma, como rectorragia, anemia, dolor abdominal, constipación y diarrea. Se recomiendan además procedimientos endoscópicos desde los 15 años, incluso antes si existen síntomas.5
La cirugía profiláctica es considerada en pacientes que presentan: poliposis colorrectal no tratable por endoscopía (más de 50-100 pólipos), síntomas severos de hemorragia gastrointestinal o diarrea; pólipos juveniles displásicos, y pacientes con historia familiar de cáncer colorrectal. Las cirugías propuestas son la colectomía subtotal con anastomosis ileorrectal o la proctocolectomía total.6
Conclusiones
Se trata de un adolescente con antecedente de poliposis y dos intervenciones quirúrgicas previas por abdomen agudo obstructivo, a quien se le realizó endoscopía digestiva alta y colonoscopía con varias polipectomías cuya histopatología dio como resultado pólipos juveniles. Posterior a esto, presenta cuadro de sangrado digestivo bajo acompañado de un abdomen agudo obstructivo, por lo que se somete a una laparotomía exploratoria en la que se encuentra una intususcepción. Se realiza resección intestinal con una entero-entero anastomosis latero-lateral de intestino delgado, con evolución favorable.
Conflictos de intereses. No existen conflictos de intereses.
Sostén financiero. Propio de los autores.
Referencias
- U.S. National Library of Medicine. Genetics Home Reference Juvenile polyposis syndrome. [Online] 2019. Disponible en: https://ghr.nlm.nih.gov/condition/juvenile-polyposis-syndrome.
- Cohen S, Hyer W, Mas E, Auth M, Attard TM, Spalinger J, et al. Management of Juvenile Polyposis Syndrome in Children and Adolescents: A Position Paper From the ESPGHAN Polyposis Working Group. Journal of Pediatric Gastroenterology and Nutrition 2019; 68 (3): 453-462.
- Cázares-Méndez JM, Zamudio-Vázquez VP, Gómez-Morales E, Ortiz-Aguirre SG, Cadena-León JF, Toro-Monjaraz EM, y cols. Pólipos gastrointestinales en pediatría. Acta Pediátrica de México 2015; 36 (1): 158-163.
- Caserras XB, Urturi CÁ. Poliposis colorrectales poco frecuentes. Gastroenterología y Hepatología Continuada 2010; 9 (2): 60-63.
- Haidle JL, Howe JR. Juvenile Polyposis Syndrome. [Online] 2003. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1469/
- Brosens LA, Langeveld D, Hattem WAV, Giardiello FM, Offerhaus GJA. Juvenile polyposis syndrome. World J Gastroenterol 2011; 17 (44): 4839-4844.
- Tam B, Salamon Á, Bajtai A, Németh A, Kiss J, Simon L, et al. The real face of juvenile polyposis syndrome. J Gastrointest Oncol 2012; 3 (4): 362-368.
- Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. American Journal of Gastroenterology 2015; 110 (2): 223-262.
- LelliJr JL. Polypoid Diseases of the Gastrointestinal Tract. In Caldamone A, Adzick NS, Krummel TM, Laberge JM, Coran RSAG. Pediatric Surgery. Filadelfia: Elsevier 2012; séptima edición: 848.
- Chow E, Macrae F. A review of juvenile polyposis syndrome. Journal of Gastroenterology and Hepatology 2005; 20 (11): 1634-1640.
- Cichy W, Klincewicz B, Plawski A. Juvenile polyposis syndrome. Archives of Medical Science 2014; 10 (3): 570-577.
- Flybase.org. Human Disease Model Report: juvenile polyposis syndrome. [Online] 2019. Disponible en: https://flybase.org/reports/FBhh0000897.html
- Jackson SB, Villano NP, Benhammou JN, Lewis M, Pisegna JR, Padua D. Gastrointestinal Manifestations of Hereditary Hemorrhagic Telangiectasia (HHT): A Systematic Review of the Literature. Digestive Diseases and Sciences 2017; 62 (10): 2623-2630.
- Aytac E, Sulu B, Heald B, O’Malley M, LaGuardia L, Remzi F, et al. Genotype-defined cancer risk in juvenile polyposis syndrome. British Journal of Surgery 2015; 102 (1): 114-118.
- Latchford AR, Neale K, Phillips RK, Clark SK. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Diseases of the Colon & Rectum 2012; 55 (10): 1038-1043.
- Gorlin RJ, Cohen MM Jr, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. American Journal of Medical Genetics 1992; 44 (3): 307-314.
- Hornick JL, Odze. RD. Polyps of the Large Intestine. In Goldblum JR, Odze RD. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. Elsevier 2009; segunda edición: 481-533.
- Feely M, González R. Juvenile polyposis syndrome. [Online] 2019. Disponible en: https://www.pathologyoutlines.com/topic/colontumorjuvenilepolyposissx.html
- Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Annals of Surgical Oncology 1998; 5 (8): 751-756.
- Brosens LAA, Hattem Av, Hylind LM, Iacobuzio-Donahue C, Romans KE, Axilbund J, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007; 56 (7): 965-967.
Correspondencia: Rubén Muñoz Cedeño
Calle 32 entre Nicolás Augusto González y Rosendo Avilés.
Guayaquil, Ecuador.
Correo electrónico: drubenmunoz@gmail.com
Acta Gastroenterol Latinoam 2020;50(4):479-484
