Neith Ortega Pérez, David Fernández San Millán, Dácil Montesdeoca Cabrera, Esteban Pérez Alonso, Juan Ramon Hernández Hernández
Servicio de Cirugía General y de Aparato Digestivo, Complejo Universitario Hospital Insular Materno Infantil. Las Palmas de Gran Canaria, España.
Acta Gastroenterol Latinoam 2019;49(3):250-253
Recibido: 09/04/2018 / Aprobado: 31/05/2018 / Publicado en www.actagastro.org el 17/09/2019

Resumen
La fibromatosis mesentérica es una entidad rara y se define como una neoplasia monoclonal derivada de estructuras muscoloaponeuróticas. A pesar de su comportamiento benigno muestra un alto riesgo de recurrencia local y de crecimiento agresivo. El tipo de tratamiento a elegir resulta una decisión difícil debido a su presentación inespecífica. Presentamos el caso de una fibromatosis mesentérica que se manifestó clínicamente como un abdomen agudo y revisamos algunos aspectos de este tumor.
Palabras claves. Fibromatosis mesentérica, poliposis adenomatosa familiar, tumores desmolde.
Acute abdomen as presentation of mesenteric fibromatosis
Summary
Mesenteric fibromatosis is a rare condition defined as a monoclonal neoplasia derived from musculoaponeurotic structures. Despite its benign nature, shows a high risk of local recurrence and aggressive growth. The type of treatment to apply is a difficult decision due to non-specific presentation. Here we present a case of a mesenteric fibromatosis which clinically showed as an acute abdomen and we review some aspects of this tumor.
Key words. Mesenteric fibromatosis, familial adenomatous polyposis, desmoid tumors.
Abreviaturas
FM: fibromatosis mesentérica.
PAF: poliposis adenomatosa familiar.
GIST: tumor del estroma gastrointestinal.
TD: tumor desmoide.
La fibromatosis mesentérica (FM) es una forma rara de proliferación fibroblástica de características histológicas benignas, con crecimiento local rápido, que afecta fundamentalmente al mesenterio del intestino delgado.1
Según la localización se dividen en abdominal (pared abdominal anterior), extraabdominal (tórax, extremidades, cabeza y cuello) e intraabdominal (mesenterio, pelvis, intra o retroperitoneal), representando estos últimos aproximadamente un 50% del total. Se describen dos tipos diferentes: los esporádicos y aquellos asociados a poliposis adenomatosa familiar (PAF) (5-10%). Mientras que los desmoides esporádicos abdominales son más comunes en las mujeres en edad reproductiva, los tumores desmoides extraabdominales son más comunes en varones y los intraabdominales se relacionan con PAF asociados al síndrome de Gardner.2
Caso clínico
Presentamos el caso de una FM que se manifestó clínicamente como un abdomen agudo. Mujer de 30 años con antecedentes personales de parálisis cerebral infantil y tetraparesia espástica, así como varias cirugías traumatológicas. Acude a urgencias por deterioro del estado general, presentando pérdida de peso, fiebre hasta 38 ºC y rictus doloroso que había empeorado en los últimos días. La exploración física fue difícil por la enfermedad de base de la paciente, pero se evidenció un mal estado general.
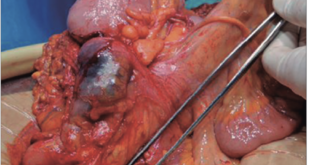
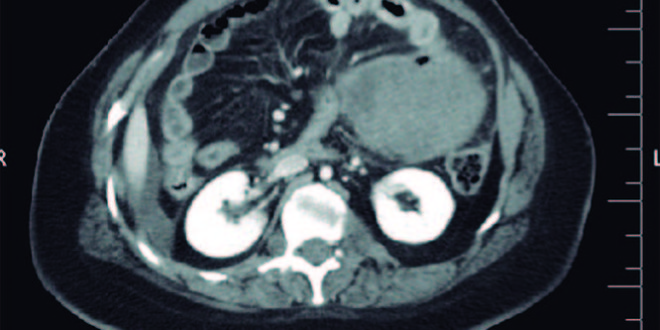
A la palpación abdominal se detecta una masa dolorosa en flanco derecho, y en la ecografía solicitada de urgencia se confirma la presencia de una tumoración de 9 x 9 x 7 cm dependiente del intestino delgado con líquido libre (Figura 1). Se realizó una laparotomía y se constató una peritonitis purulenta generalizada secundaria a tumoración perforada en íleon distal compatible macroscópicamente con tumor estromal gastrointestinal, conocido como GIST (Figura 2), por lo que se realizó resección ileocecal con anastomosis ileocólica.
El informe anatomopatológico definitivo de la pieza quirúrgica resecada confirmó el diagnóstico de FM tipo desmoide (Figura 3). El postoperatorio se desarrolló sin incidencias, y se presentó como única complicación una infección de la herida quirúrgica que se resolvió de forma satisfactoria. Actualmente la paciente se encuentra asintomática.
Figura 1. TC abdominal. Se observa la presencia de una masa que parece depender del intestino delgado y líquido libre en relación con una probable perforación tumoral.
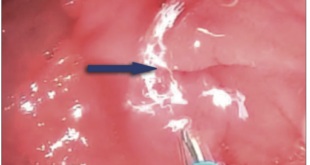
Figuras 2 y 3. Aspecto macro y microscópico de la tumoración. Suele presentar un patrón mal circunscrito que se compone de la proliferación de células fusiformes estrelladas que forman fascículos largos sin mostrar generalmente atipía nuclear o hipercromasia, que son fuertemente positivas con vimentina. La inmunorreactividad nuclear a betacatenina se expresa frecuentemente, en el 67-80% de los casos.
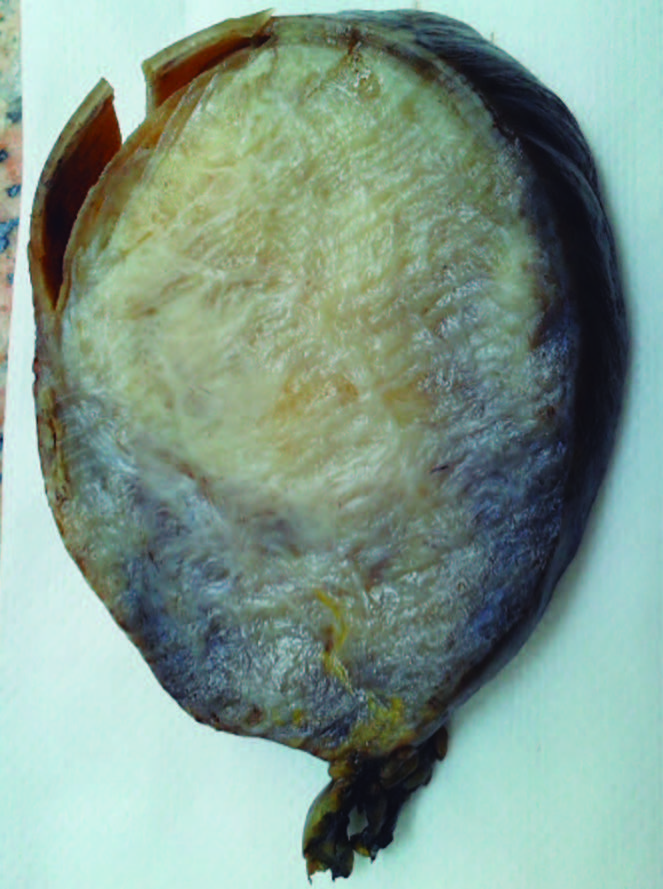
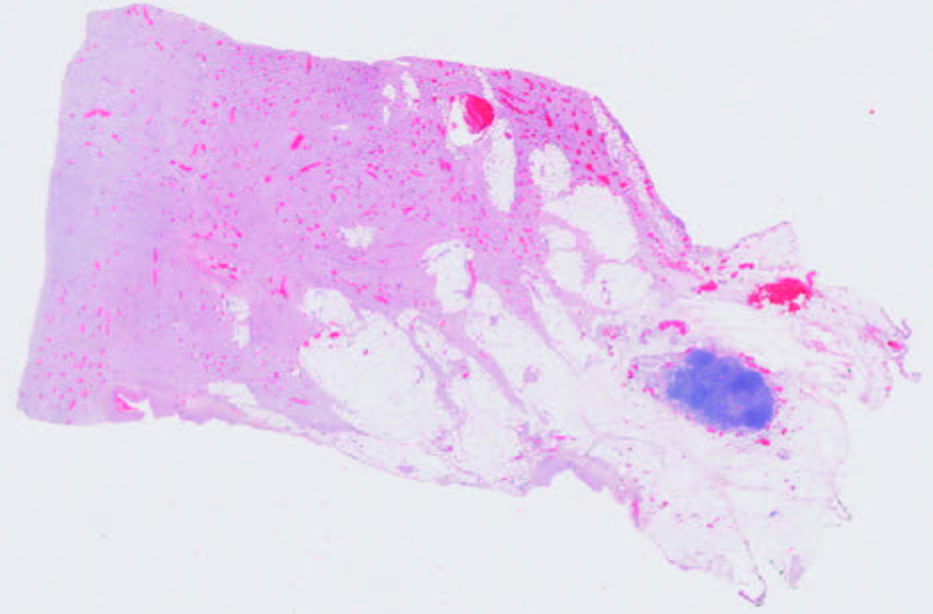
Discusión
La FM se trata de una neoplasia rara de estirpe miofibroblástica, no metastatizante, pero localmente agresiva y con tendencia a la recurrencia. Presenta una prevalencia del 3% de los tumores de partes blandas y el 0,03% de todas las neoplasias.2 El subtipo de tumor desmoide (TD) constituye la causa más frecuente de tumor primario en el mesenterio.3
El tumor aparece más frecuentemente en el mesenterio del intestino delgado, y su localización es menos frecuente en el mesocolon transverso, ligamento gastrohepático y ligamento gastroesplénico.1 Los TD se dividen en 2 grupos: esporádicos y asociados a la PAF (< 10% del total).4-8 Clínicamente el comportamiento puede ser muy variado y depende de la localización; existiendo en un extremo del espectro formas asintomáticas, de pequeño tamaño y crecimiento lento, y en el otro, lesiones rápidamente progresivas y de gran tamaño que pueden producir complicaciones por compresión u obstrucción de estructuras vecinas, como cuadro de oclusión intestinal o hidronefrosis.3, 6 La clínica más frecuente es la de una masa abdominal cuya sintomatología dependerá de su localización.1 Nuestro caso debutó como cuadro de abdomen agudo secundario a la perforación de la tumoración.
En cuanto al diagnóstico, las técnicas de imagen aportan datos anatómicos que definen la relación del tumor con las estructuras adyacentes y permiten evaluar la resecabilidad y posibilidades de tratamiento.4-6 El método diagnóstico de elección es la tomografía computada (TC), que nos confirma la heterogeneidad de la masa, la localización mesentérica y la relación con las estructuras vasculares y parenquimatosas adyacentes.1
Resulta fundamental el estudio del colon en estos pacientes, debido a la frecuente asociación de la FM con el síndrome de Gardner. En nuestro caso, la existencia de un síndrome de Gardner asociado se descartó debido a la colonoscopía normal.
El diagnóstico de confirmación es histológico y por inmunohistoquímica de la biopsia o pieza quirúrgica donde la inmunorreactividad nuclear a betacatenina se expresa frecuentemente en el 67-80% de los casos.4 Si bien histopatológicamente presenta signos de benignidad, presenta un comportamiento biológico que implica una tendencia a la infiltración de los tejidos adyacentes y a la recidiva local tras su extirpación.1, 3
El diagnóstico diferencial de los TD intraabdominales debe realizarse con el tumor del estroma gastrointestinal (GIST), el tumor fibroso solitario, el tumor inflamatorio miofibroblástico, la mesenteritis esclerosante y la fibrosis retroperitoneal (ya sea idiopática o secundaria a fármacos o a enfermedad maligna subyacente).3 Los TD abdominales pueden coexistir con los GIST, sin embargo, el diagnóstico diferencial suele resultar sencillo, ya que estos suelen expresar c-kit (mientras que en la fibromatosis este es persistentemente negativo), CD34, y son casi siempre betacatenina negativo.3
En nuestro caso no pudo establecerse un diagnóstico preoperatorio con exactitud, debido a su debut como abdomen agudo, por lo que estuvo justificada la realización de una laparotomía exploradora con fines tanto diagnóstico como terapéutico. Además, en nuestra paciente la sospecha de GIST se debía a la morfología de la tumefacción, pero los hallazgos de la anatomía patológica e inmunohistoquímica confirmaron el diagnóstico de FM tipo desmoide.
Así mismo, debemos descartar entre otras posibilidades diagnósticas el tumor carcinoide, sobre todo aquellos que se originan a nivel del tracto biliar o páncreas. El linfoma, la esclerosis mesentérica y el fibrosarcoma retroperitoneal también deberían descartarse.2, 8
La actitud terapéutica a elegir es una decisión difícil debido a que no existe consenso sobre su aplicación ni sobre las indicaciones. Las posibilidades terapéuticas comprenden la observación clínica y con métodos de imagen en aquellos pacientes con TD pequeños que no afectan a estructuras cercanas o para pacientes con síntomas leves, de forma que pueden ser seguidos clínicamente y con métodos de imagen.3, 5, 7
Sin embargo, el tratamiento de elección lo constituye la extirpación quirúrgica completa del tumor con márgenes microscópicos negativos, con preservación de la función orgánica provocando mínima morbilidad.3, 5 Debido a la capacidad de infiltración local es frecuente la extirpación incompleta con la consecuente aparición de recidivas locales, de ahí que se hayan desarrollado diferentes tratamientos adyudantes.1 Entre ellos tenemos la radioterapia, que constituye una opción inicial eficaz para pacientes no candidatos a cirugía o que la rechazan la misma o en los cuales la morbilidad quirúrgica sería inaceptable. Pero su indicación está justificada como tratamiento adyuvante en el caso de márgenes quirúrgicos positivos, en un intento de disminuir el riesgo de recurrencia local.
En cuanto al tratamiento médico existen diferentes opciones, desde el uso de la quimioterapia citotóxica en caso de tumores irresecables o refractarios a otros tratamientos hasta el uso de antiestrógenos. Así mismo, su combinación con antiinflamatorios no esteroideos tipo sulindac o indometacina constituye otra opción terapéutica.
Como se apunta en algunos artículos el tratamiento con tamoxifeno en dosis elevadas y sulindac en pacientes con PAF y TD resulta efectivo, presentando una respues ta parcial o completa. Además, se están desarrollando en la actualidad diferentes terapias moleculares como el uso del interferón o el imantinib; este último es eficaz en la fibromatosis ya que estos tumores expresan receptores de PDGF.5 Sin embargo, ninguno de estos enfoques resulta totalmente eficaz en la mayoría de los pacientes. Ello, unido a la ausencia de estudios aleatorizados y prospectivos para el tratamiento, hace que su abordaje óptimo sea todo un reto, por lo que el tratamiento debe ser individualizado.1, 8
La historia natural de estos tumores es variable. Se ha descrito la resolución espontánea en el 10%, recurrencia en el 30%, el 50% se mantienen estables después del diagnóstico y el 10% son de progresión rápida.2
En nuestro caso, como ya se ha comentado, la paciente tuvo que ser intervenida de forma urgente y en la actualidad se encuentra asintomática sin recibir tratamiento adyuvante y sin hallazgos clínicos ni radiológicos que nos hagan sospechar recidiva.
Así mismo, estos pacientes deberán someterse a un seguimiento estricto clínico y radiológico durante los primeros años debido a la elevada tasa de recidiva local.8
Conflicto de intereses. Los autores declaran no tener ningún conflicto de intereses.
Sostén financiero. No se recibió apoyo financiero para la realización de este manuscrito.
Referencias
- Fernández Bermejo M, Santander Vaquero C, Olivera J, Lo Iacono O, García Buey L, Pajares García JM. Fibromatosis mesentérica como causa de masa abdominal única. Gastroenterol Hepatol 2000; 23: 126-128.
- Luzón Solanas L, Montoro Huguet M. Enfermedades del mesenterio. Procesos inflamatorios. Patología vascular. Isquemia mesentérica. Medicine 2016; 12: 178-188.
- Vida Pérez L, Martínez Rivas F. Tumores desmoides intraabdominales. Med Clin (Barc) 2013; 141: 314-319.
- Quintini C, Ward G, Shatnawei A, Xhaja X, Hashimoto K, Steiger E, Hammel J, Diago Uso T, Burke CA, Church JM. Mortality of intra-abdominal desmoid tumors in patients with familial adenomatous polyposis: a single center review of 154 patients. Ann Surg 2012; 255; 511-516.
- Vasen HF, Möslein G, Alonso A, Aretz S, Bernstein I, Bertario L, Blanco I, Bülow S, Burn J, Capella G, Colas C, Engel C, Frayling I, Friedl W, Hes FJ, Hodgson S, Järvinen H, Mecklin JP, Moller P, Myrhoi T, Nagengast FM, Parc Y, Phillips R, Clark SK, de Leon MP, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Tejpar S, Thomas HJ, Wijnen J. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57: 704-713.
- González MA, Menéndez R, Ayala JM, Herrero M, Cuesta J, Domínguez A, Martínez M, Graña JL, Pozo F. Intra-abdominal desmoid tumor. Cir Esp 2005; 77: 362-364.
- Reddy Kallam A, Vrama Krishna B, Kishore Roy G, Karthik KRV. Desmoid Tumours: Our Experience of Six Cases and Review of Literature. Journal of Clinical and Diagnostic Research 2014; 8: NE01-NE04.
- Galletto P, Leoz ML, Castells A, Balaguer F. Tumores desmoides intraabdominales en la poliposis adenomatosa familiar. Gastroenterol Hepatol 2013; 36: 580-586.
Correspondencia: Neith Ortega Pérez
Avenida Marítima del Sur, s/n (35016). Las Palmas de Gran Canaria, España
Tel.: 928444000/690651747
Correo electrónico: neithortega@gmail.com
Acta Gastroenterol Latinoam 2019;49(3): 250-253