Mirta Ciocca,1 Cinthia Bastianelli,2 Patricia Nacif,3 Gilda Porta,4 Flor Ramírez,5 Gloria Ríos,6 Jorge Romero,7 Carolina Rumbo8
1 Hospital Alemán. Ciudad Autónoma de Buenos Aires, Argentina.
2 Hospital Materno Infantil de Salta. Salta, Argentina.
3 Centro Hospitalario Pereira Rossell. Montevideo, Uruguay.
4 Hospital Sirio-Libanes A C Camargo Cáncer Center. San Pablo, Brasil.
5 Hospital Nacional San Juan de Dios. Catedrática Asociada de Pediatría de la Universidad de San Carlos. Ciudad de Guatemala, Guatemala.
6 Clínica Alemana. Santiago de Chile, Chile.
7 Hospital Infantil Juan Pablo II y Sanatorio Nuestra Señora del Pilar. Ciudad de Guatemala, Guatemala.
8 Instituto de Trasplante Multiorgánico. Hospital Universitario Fundación Favaloro. Ciudad Autónoma de Buenos Aires, Argentina.
Acta Gastroenterol Latinoam 2016;46: 237-245
Recibido: 13/10/2015 / Aprobado: 24/04/2016 / Publicado en www.actagastro.org el 03/10/2016

Resumen
La hepatitis autoinmune es un proceso inflamatorio hepático crónico, de etiología desconocida, que predomina en el sexo femenino. Librado a su evolución espontánea, puede progresar a cirrosis en la mayoría de los casos. Sus manifestaciones clínicas de comienzo son variables, desde una forma asintomática hasta una hepatitis severa con insuficiencia hepática aguda. Es una enfermedad compleja, poligénica, multifactorial, originada por la interacción de factores ambientales y causales en individuos con predisposición genética a desarrollar enfermedades autoinmunes. Se caracteriza bioquímica y serológicamente por la presencia de transaminasas elevadas, hipergammaglobulinemia y autoanticuerpos circulantes, e histológicamente por hepatitis de interfase. La presencia de autoanticuerpos tiene valor diagnóstico y nos permite distinguir 2 tipos de hepatitis autoinmune: tipo 1, con anticuerpos anti-músculo liso, anti-actina y/o anti-núcleo y/o antígeno soluble hepático y tipo 2 con anticuerpos anti-microsomas de hígado y riñón o anti-citosol hepático. El primer esquema terapéutico implementado consistió en la asociación de prednisona y azatioprina, que resulta beneficioso en el 80% de los casos. Alrededor del 20% de los pacientes no responden al tratamiento convencional, requiriendo un tratamiento alternativo para impedir la progresión de la enfermedad. El trasplante hepático está indicado en aquellas hepatitis autoinmunes que se presentan como insuficiencia hepática aguda, o en aquellos pacientes en los que la enfermedad progresa a insuficiencia hepática terminal, a pesar del tratamiento instituido (10-20%).
Palabras claves. Hepatitis autoinmune, infancia, enfermedad hepática, tratamiento.
Autoimmune hepatitis in childhood. Working group of the Latinamerican Society of Pediatric Gastroenterology, Hepatology and Nutrition (LASPGHAN)
Summary
Autoimmune hepatitis is an inflamatory, chronic hepatic process of unknown etiology, affecting mainly females. Due to its spontaneous evolution, if not treated, it may progress to cirrosis in most cases. The clinical features at the onset of the disease vary from asymptomatic to severe hepatitis and liver failure. It is a complex, polygenic, multifactorial disease, produced by the interplay of environmental and etiological factors in patients with a genetic predisposition to develop autoimmune diseases. The biochemical and serological features are elevated transaminase, hypergammaglobulinemia and circulating autoantibodies, and histologic picture of interface hepatitis. According to serum autoantibodies on diagnosis, two types of autoinmune hepatitis are found: type 1 positive for antinuclear (ANA) and/or smooth muscle antibodies (SMA), and/or soluble liver antigen (SLA), and type 2 with anti-liver kidney microsomal type 1 (anti-LKM 1) and/or anti-liver cytosol type 1 antibodies (anti-LC1). Treatments used for autoinmune hepatitis include prednisone and azathioprine, being effective in 80% of patientes. Almost 20% of patients do not react to conventional treatment, requiring another alternative therapy preventing the progression of the disease. Liver transplantation is indicated in patients with acute liver failure or those patients who develop end-stage liver disease.
Key words. Autoimmune hepatitis, childhood, liver disease, treatment.
Introducción
La hepatitis autoinmune (HAI) es un proceso inflamatorio hepático crónico, de etiología desconocida, que librado a su evolución espontánea puede progresar a cirrosis y enfermedad hepática terminal. Predomina en el sexo femenino y se caracteriza bioquímica y serológicamente por la presencia de transaminasas elevadas, hipergammaglobulinemia y autoanticuerpos circulantes, e histológicamente por hepatitis de interfase. Es fundamental el diagnóstico temprano, ya que el tratamiento inmunosupresor puede salvar la vida del paciente.1-6
Clasificación
De acuerdo con los autoanticuerpos presentes en el suero al momento del diagnóstico, se distinguen 2 tipos de HAI:
- HAI Tipo 1: asociada a autoanticuerpos antinucleares (ANA) y/o antimúsculo liso (smooth-muscle-antibody, SMA), cuya especificidad es mayor si se comprueba que son antiactina y/o antígeno soluble hepático (soluble liver antigen, SLA).
- HAI Tipo 2: asociada a anticuerpos anti-microsomas de hígado y riñón tipo 1 (liver-kidney-microsome-antibody, anti-LKM 1) y/o anti-citosol hepático tipo 1 (liver cytosol, anti-LC1).
El 40% de la HAI tipo 1 y el 80% de la tipo 2 se diagnostican antes de los 18 años de edad (edades promedio 10 y 6,5 años respectivamente). Aunque la mayor prevalencia ocurre en el rango de edad prepuberal, se ha diagnosticado HAI en edades tan tempranas como los 6 meses de vida. Esta enfermedad se observa más frecuentemente en el sexo femenino, con una relación 4:1 para la HAI tipo 1 y 9:1 para la tipo 2.1-5
Patogenia
La HAI es una enfermedad compleja, multifactorial, poligénica, causada por la interacción de factores ambientales con otros causales, en personas con una predisposición genética a desarrollar enfermedades autoinmunes.6
Variantes o polimorfismos genéticos específicos aumentan o disminuyen el riesgo de padecer enfermedades autoinmunes, afectando el fenotipo clínico como ocurre en la HAI tipo 1.7, 8 En la HAI tipo 2, la autorreactividad contra autoantígenos específicos, tales como el citocromo P450IID6 (CYP2D6), es la base patogénica de esta enfermedad.9
El mecanismo que conduce a la destrucción celular y ruptura de la autotolerancia inmunológica es desconocido aunque se sabe que hay una pérdida de la regulación inmune. Se cree que la injuria hepática se produce en respuesta a la presentación de un péptido autoantigénico al receptor de un linfocito T (Th0) mediante las células presentadoras de antígeno (APC) que pueden ser macrófagos, células dendríticas y linfocitos B, en presencia de señales coestimuladoras inducidas por la interacción de los linfocitos CD28 sobre Th0 y los CD80 sobre las APC. El daño hepático está orquestado por los linfocitos CD4 + que reconocen un antígeno propio, que es un péptido ubicado en las APC.10
Para desencadenar la respuesta autoinmune, el péptido se acopla a una molécula de antígeno de histocompatibilidad (HLA) de clase II y es presentado a las células CD4 + Th0 que se activan y se diferencian en Th1 y Th2 en presencia de IL-12 o IL-4, y células TH17 cuando hay un predominio de IL-6. Para desencadenar la respuesta autoinmune, el péptido está acoplado a una molécula de HLA de clase II y presentado a las células Th0, ocurriendo una interacción entre las dos células. Las células Th0 se activan y se diferencian en Th1 y Th2, e inician una cascada de eventos inmunológicos. Los macrófagos presentes en el hígado producen IL-12, las células Th1 secretan principalmente IL-2 e IF-γ, que a su vez activan más macrófagos, aumentan la expresión de moléculas HLA de clase I, incrementándose la vulnerabilidad de los hepatocitos al ataque citotóxico, e inducen la expresión de HLA de clase II sobre los hepatocitos, siendo capaces de presentar el autoantígeno (péptido) a las células Th1, perpetuando de esta forma el ciclo de reconocimiento inmune. Las células Th2, en presencia de un microambiente rico en IL-4, producen IL-4, IL-5 e IL-10, induciendo la producción de autoanticuerpos a partir de linfocitos B y plasmocitos activados. Una vez desencadenada la reacción autoinmune, los hepatocitos son destruidos por diversos mecanismos: directamente en presencia de HLA de clase I, por intermedio de la citotoxicidad de los linfocitos CD8 +, lisis por la acción de citoquinas o por los autoanticuerpos ligados al complemento o por las células NK (asesinas naturales en castellano y natural killer en inglés). Los hepatocitos cubiertos por los autoanticuerpos pueden ser destruidos por la acción del complemento o por el receptor Fc, dos anticuerpos unidos a los linfocitos NK. El proceso de reconocimiento autoantigénico está estrictamente controlado por mecanismos de regulación representados por las células T reguladoras (T-regs) CD4 + y CD25 +.11-14
Una pérdida en las células T-regs conduce a la pérdida de tolerancia inmunológica en HAI y respuestas efectoras inmunes descontroladas.15 Las células T-regs expresan diversos marcadores incluyendo el factor de receptor de TNF inducido por glucocorticoides (GITR, glucocorticoid-induced TNFR family related gene), CTLA- 4 (antígeno 4 del linfocito T citotóxico) y FOXP3. Hay un aumento en la expresión de FOXP3 en la HAI y las células T-regs pueden estar disminuidas en número o reducida su función, generando una interrupción en la modulación de la proliferación de células Th2, con aumento de la producción de citoquinas, lo cual facilita el daño hepático. Además, las células T-regs aumentan la activación de los monocitos, células del sistema inmune innato que abundan en el infiltrado inflamatorio periportal.15-18 Los corticosteroides pueden reconstruir la función de las células T-regs y atenuar la respuesta celular citotóxica inmunomediada.
Existe una predisposición genética en la HAI vinculada con los HLA. Variaciones geográficas y los diferentes subtipos de HLA contribuyen a diferentes riesgos relativos de los diferentes grupos étnicos. En Europa y EE.UU., los haplotipos HLA DRB1 / 0301 y DRB1 / 0401 están fuertemente asociados con HAI tipo 1, mientras que la presencia de DR1501 parece ser protectora.19, 20
En Japón domina la fuerte asociación con el haplotipo HLA DR0405, mientras que en América del Sur (Argentina y Brasil) hay un predominio de HLA DR1301 y en menor proporción con DR3 o DR4. La asociación DR7 con HAI tipo 2 se describe en todos los grupos étnicos.21-23
Las deficiencias de IgA y del complemento 4a, condicionadas genéticamente, pueden estar asociadas a HAI.24 La deficiencia de IgA es más frecuente en los pacientes con HAI tipo 2 y está genéticamente vinculada a HLA-DR1 y DR7. Se han observado bajos niveles de complemento 4a en la población pediátrica.25
Otros genes no relacionados con el HLA pueden estar involucrados en la susceptibilidad a la HAI. El antígeno 4 del linfocito T citotóxico (CTLA-4), también conocido como CD152, es una molécula presente en la superficie de los linfocitos T que interactúa compitiendo con los CD28 y los ligandos B7-1 y B7-2 a las APC, transmitiendo una señal inhibidora a las células T. El cambio de adenina (A) por guanina (G) en el exón 1 del gen CTLA- 4 confiere susceptibilidad a algunas enfermedades autoinmunes, incluyendo la HAI en individuos caucásicos en los EE.UU.8 Estos datos no se encontraron en pacientes brasileños con HAI.26 En pacientes de Europa y EE.UU. con HAI tipo 1, se encontró polimorfismo en la posición 308 del promotor del gen del TNFα, en comparación con individuos sanos, asociándoselo con peor respuesta terapéutica. Estos datos no fueron confirmados en pacientes japoneses. Otro polimorfismo en la posición 670 en el promotor del gen FAS se asocia con una progresión de la enfermedad más agresiva.27 Los polimorfismos de los receptores de vitamina D se asocian en la HAI con la activación de los macrófagos, impidiendo la diferenciación de células dendríticas e inhibiendo la función de las células Th1.28
En el 20% de los pacientes con HAI tipo 2 puede asociarse la poliendocrinopatía autoinmune tipo 1 o síndrome APECED (candidiasis crónica mucocutánea, hipoparatiroidismo e insuficiencia adrenal autoinmune). Es una enfermedad autosómica recesiva con múltiples enfermedades autoinmunes órgano-específicas. La enfermedad es causada por mutaciones en el gen AIRE (21q22.3) que codifica para el factor de transcripción AIRE, el cual está implicado en los mecanismos de tolerancia inmune y contribuye a la selección negativa de los linfocitos T autorreactivos en el timo, los ganglios linfáticos y el bazo.29
Diagnóstico
Manifestaciones clínicas
La HAI tipo 2 es más común en niños que en adultos y a pesar de compartir la mayoría de los síntomas clínicos de la HAI tipo I, tiende a ser más severa, manifestándose frecuentemente como un proceso agudo y con mayor tendencia a una rápida progresión a la cirrosis.
Describir un perfil clínico específico es difícil, por la naturaleza fluctuante de la enfermedad. Aproximadamente el 50% de los niños con HAI presentan un cuadro de enfermedad aguda con ictericia, coluria, fiebre, astenia, anorexia y aumento del volumen abdominal. En el 10-15% el comienzo puede ser insidioso, con síntomas inespecíficos como fatiga, náusea, dolor abdominal, artralgias, amenorrea primaria o secundaria. Sin embargo, el espectro clínico es amplio, desde presentaciones asintomáticas a un cuadro agudo severo de insuficiencia hepática aguda.
Al examen físico puede observarse hepatomegalia con o sin esplenomegalia y estigmas de hepatopatía crónica. Ocasionalmente el niño puede presentar ascitis. Los niveles de bilirrubinas séricas usualmente están elevados, así como las aminotransferasas.
La HAI debe sospecharse en todo niño con aparente cuadro de hepatitis aguda, de etiología desconocida, que no presenta mejoría luego de las 4-6 semanas del comienzo de los síntomas. La presentación como insuficiencia hepática aguda suele ser más común en la HAI tipo 2.
Además, los niños pueden presentarse con un proceso crónico descompensado que se asemeja a una insuficiencia hepática aguda (ictericia severa, coagulopatía, encefalopatía), pero asociado a ascitis, circulación colateral y otros elementos clínicos característicos de un proceso crónico, especialmente si el diagnóstico no se ha hecho en una fase temprana.
El efecto de las manifestaciones clínicas tiene impacto en la calidad de vida de los pacientes. Gulati y col realizaron una relación del indicador de calidad de vida vinculado con la salud de un grupo de pacientes con HAI. Este estudio demostró que la fatiga, el dolor abdominal y los aspectos psicológicos intrínsecos eran los elementos que afectaban en mayor medida el diario vivir de estos pacientes.1-6, 30
Laboratorio
Si bien no existe ninguna alteración bioquímica específica en la HAI, los hallazgos característicos son:
- Elevación de los niveles de transaminasas entre 2 y 50 veces del valor normal.
- La hipergammaglobulinemia de tipo IgG, que suele ser mayor al 1,5 del valor normal. En la infancia se ha demostrado correlación entre los niveles de IgG y el grado de actividad de la enfermedad. En un 10 a un 20% de los niños puede ser normal.
- La hiperbilirrubinemia y los niveles normales de gamma glutamil transpeptidasa o levemente aumentados, son hallazgos frecuentes.
- El déficit de IgA por debajo de 1,2 gr/l se presenta en el 45% de la HAI tipo 2 y en el 9% de la HAI tipo 1.
- Los niveles de C4 pueden estar disminuidos hasta en el 69% de los casos.
- Leucopenia y pancitopenia se observan en pacientes con cirrosis e hipertensión portal, secundaria a hiperesplenismo.
- La hipoalbuminemia y el déficit de los factores de la coagulación se presentan en los niños con insuficiencia hepática.
- La coagulación puede ser anormal principalmente en la enfermedad crónica avanzada o hepatitis fulminante.1-6, 31-35
Autoanticuerpos
En la HAI los autoanticuerpos ayudan al diagnóstico, pero hay que tener en cuenta que títulos bajos de los mismos e incluso negativos en una determinación, no excluyen el diagnóstico de la enfermedad (Tabla 1).
Tabla 1. Autoanticuerpos en el diagnóstico de hepatitis autoinmune.
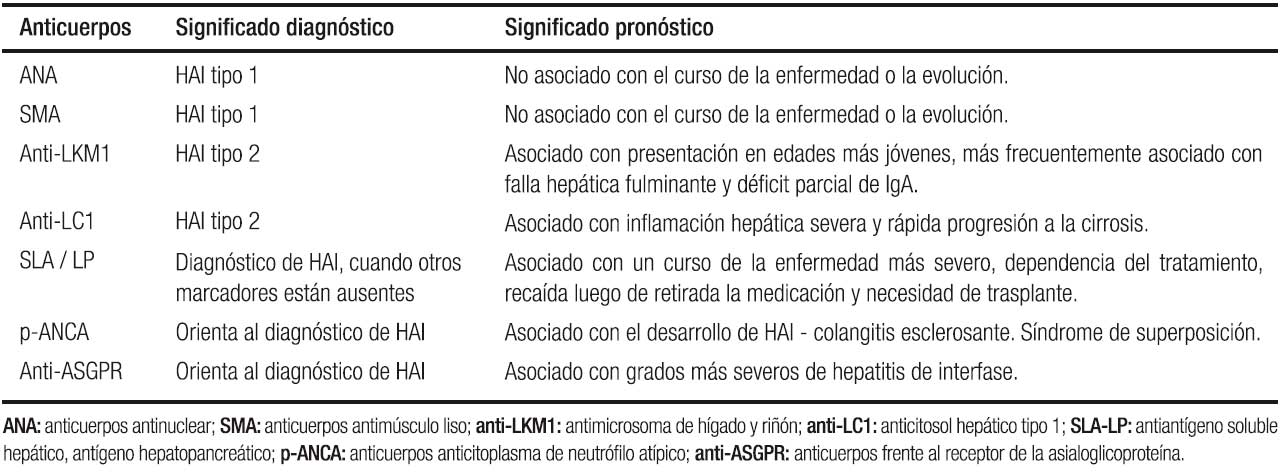
Los autoanticuerpos circulantes tienen valor diagnóstico y permiten clasificar la HAI en 2 subtipos.
HAI tipo 1
Anticuerpos antinucleares
Se determinan por inmunofluorescencia indirecta (IFI) en secciones de tejidos de riñón, hígado y células gástricas, frente al centrómero, histonas y ribonucleoproteínas. En la mayoría de los casos el patrón es homogéneo y en un porcentaje pequeño puede ser granular o moteado. Está asociado al alelo HLA DRB1 *0401. En el niño se consideran positivos valores igual o mayor a 1/20. Si bien puede encontrarse de manera aislada, suelen asociarse a la presencia de SMA.
Anticuerpos antimúsculo liso
Frente a componentes de actina, tubulina y filamentos intermedios, son detectados en secciones de riñón, estómago e hígado. En riñón tiñe vasos (V), glomérulos (G) y túbulos (T); los patrones VG y VGT son frecuentes en la HAI. A pesar de ser menos prevalentes que los ANA, son más específicos, siendo ésta mucho mayor si se comprueba que son antiactina. En el niño se consideran positivos valores igual o mayores a 1/20.
Se asocian al haplotipo HLA DR3 y tienen cierto carácter pronóstico (peor respuesta al tratamiento). Títulos igual o mayor a 1/320 de SMA casi siempre reflejan la presencia antiactina. El 20% de los pacientes SMA positivos son antiactina negativos, por lo tanto, la ausencia de reactividad antiactina no excluye la HAI tipo 1.
Los anticuerpos ANA y SMA no son específicos de la HAI tipo 1 y pueden encontrarse en otras enfermedades como la hepatitis B y C crónicas, enfermedad de Wilson, niños obesos, etc.1-6, 31-34
HAI tipo 2
Anticuerpos antimicrosomas de hígado y riñón tipo 1
Los anti-LKM1 tiñen el citoplasma de los hepatocitos y el túbulo proximal renal.
Son los principales anticuerpos detectados en la HAI tipo 2, altamente específicos y van dirigidos contra el citocromo P450. En niños se considera positivo cualquier valor igual o mayor de 1/10. Se han encontrado presentes en pacientes con hepatitis C crónica, en menos del 5%.
Anticuerpos anticitosol hepático tipo 1
Están dirigidos contra la proteína citosólica hepática. Los anti-LC1 pueden acompañar a los anticuerpos anti-LKM1 en la HAI tipo 2, e incluso pueden ser detectados como marcador aislado hasta en un 15% de los pacientes.
Se ha sugerido que su presencia se asocia con la aparición de otras enfermedades autoinmunes, mayor inflamación hepática y una rápida progresión a la cirrosis. No está claro aún si sus títulos se correlacionan con el nivel de actividad de la enfermedad.1-6, 31-34
Anticuerpos antiantígeno soluble hepático/antígeno hepatopancreático (SLA/LP)
Frente a citoqueratinas 8 y 18 y glutation S-transferasa y el LP frente a proteínas citosólicas. Presentes hasta en el 60% de los niños con HAI tipo 1. Son del subtipo IgG 1, por lo que se sugiere pueden tener su origen en ciertos estímulos inmunoespecíficos como alguna proteína viral. Parecen mostrar asociación positiva con el alelo HLA DRB1 *0301 y negativa con el HLA DRB1 *04.
Anticuerpos anticitoplasma de neutrófilos atípicos (p- ANCA)
Estos anticuerpos presentan un patrón perinuclear atípico, periférico, por IFI y parecen estar dirigidos frente a una proteína de superficie de 50 Kd. Se observan en pacientes con HAI tipo 1. Estos anticuerpos también se detectan en pacientes con colangitis esclerosante primaria y en la enfermedad inflamatoria intestinal, por lo que debe realizarse el diagnóstico diferencial con ellas.
Anticuerpos frente al receptor de la asialoglicoproteína (ASGP-R)
Están dirigidos contra una glicoproteína transmembrana de la superficie del hepatocito y probablemente tengan importancia pronóstica. Se encuentran hasta en 1/3 de los niños con HAI y pueden coexistir con ANA, SMA y LKM1. Su presencia se correlaciona con la actividad histológica y su desaparición con la respuesta al tratamiento. Su persistencia se ha asociado con probable recaída tras retirar los corticoides. No son específicos de la enfermedad y pueden estar presentes en hepatitis virales, hepatitis inducidas por drogas y en colangitis esclerosante primaria.
Los anticuerpos ANA, SMA y LKM1 son prácticamente excluyentes entre sí; en los casos excepcionales en que están presentes simultáneamente el niño debe clasificarse como HAI tipo 2.1-6, 31-34
Hallazgos histológicos
Si bien los cambios histológicos de un niño con HAI no son patognomónicos, la biopsia hepática es uno de los pilares en su diagnóstico. Permite caracterizar el compromiso hepático, evaluar el grado de daño del mismo y a la vez descartar otras etiologías. En el diagnóstico histológico es importante el tamaño de la muestra. En general, cuando se trata de una enfermedad difusa se ha planteado que la muestra debe medir 1,5 cm de largo, incluyendo al menos 6 espacios porta. Estudios en adultos con hepatitis crónica han dado cuenta de que muestras pequeñas hacen un sub-diagnóstico del daño hepático, recomendando que la muestra mida 2 cm y contenga 6-8 espacios porta.5, 35, 37-39
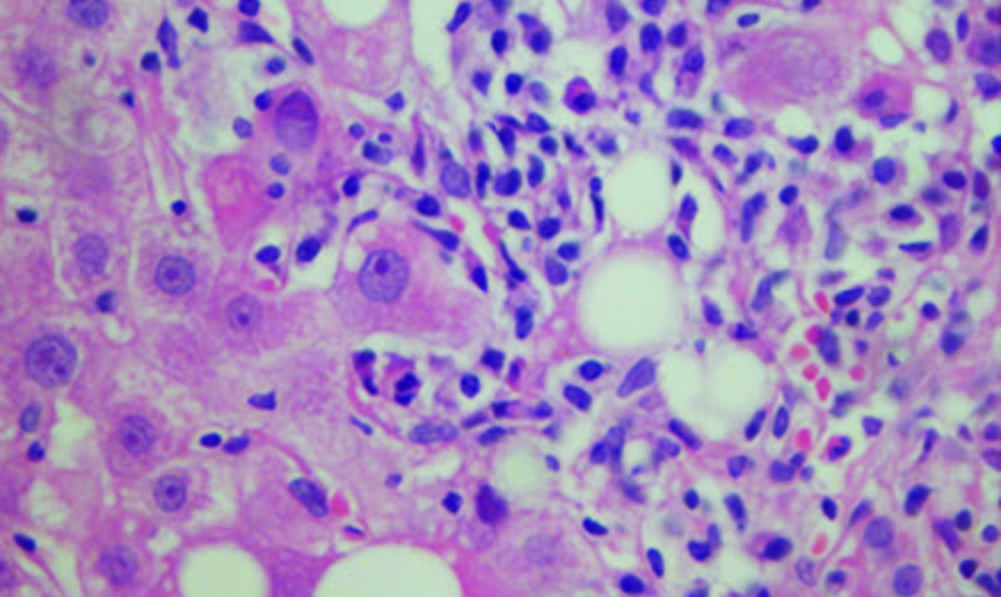
El compromiso histológico de la HAI se caracteriza por un infiltrado de linfocitos, células plasmáticas y eosinófilos a nivel del espacio porta, con difusión hacia el lóbulo hepático (zona 2 y 3), infiltración de los hepatocitos de la periferia y erosión de la placa limitante, que es lo que se ha llamado hepatitis de interfase. También se ha descrito como frecuente el hallazgo de emperipolesis, que es la presencia de linfocitos o células plasmáticas intactas dentro de un hepatocito. Por la destrucción y muerte de hepatocitos se observa colapso del tejido conectivo. La regeneración hepática da origen a la formación de rosetas (hepatocitos rodeando un canalículo biliar en el área periportal). La severidad de la hepatitis de interfase es similar en la HAI tipo 1 y 2, sin embargo, la cirrosis es más frecuente en la tipo 1. La presencia de pocas células plasmáticas no descarta la enfermedad.5, 40, 41
Cuando la HAI se presenta como insuficiencia hepática aguda, el hallazgo más característico es el compromiso necroinflamatorio centrolobular. Sin embargo, no hay consenso en los hallazgos histológicos de esta forma clínica grave de la enfermedad.
El diagnóstico diferencial histológico con daño hepático por drogas puede ser difícil; en ambos casos se puede observar hepatitis de interfase, emperipolesis y formación de rosetas, pero son más severos en HAI; al igual que infiltrado de eosinófilos en áreas portales e intra-acinar. Los linfocitos intra-acinar y el daño colestásico son más frecuentes en el daño hepático por drogas. En HAI se encuentra más necrosis focal, confluente y fibrosis que en la causa tóxica. La otra patología autoinmune con la que se plantea diagnóstico diferencial, especialmente en las etapas iniciales, es con la colangitis esclerosante.39, 42, 43
Para objetivar el grado de actividad histológica necroinflamatoria y fibrosis se usa, al igual que en otras hepatitis crónicas, la clasificación de Ishak. Este índice de actividad va de 0-18 evaluando el grado de hepatitis de interfase, necrosis confluente, inflamación focal e inflamación portal, evaluándose además el grado de fibrosis en una escala de 0-6. Este índice también permite objetivar la evolución histológica y la respuesta a tratamiento (Figuras 1, 2 y 3).44-46
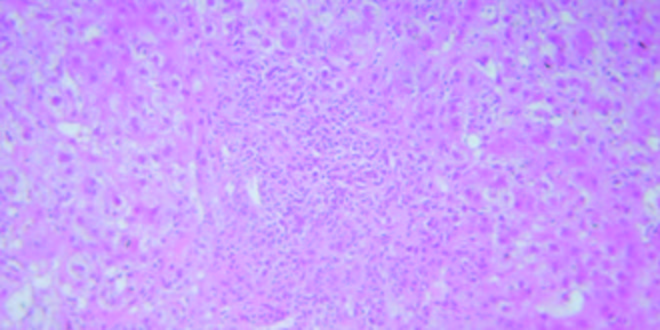
Figura 1.
Hematoxilina y Eosina 100X. Espacio porta con hepatitis de interfase linfoplasmocitaria moderada a marcada. Cortesía de los Doctores Marcelo Fabián Amante y Gabriel Casas, Servicio de Anatomía Patológica del Hospital Alemán, Buenos Aires.
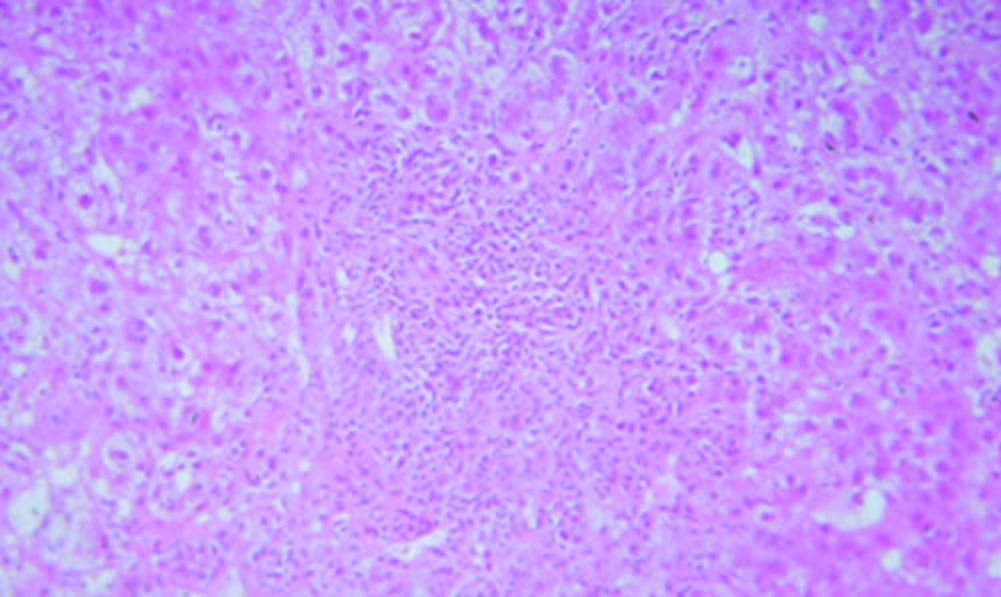
Figura 2.
Hematoxilina y Eosina 400X. Sector de interfase con inflamación linfoplasmocitaria y necrosis de hepatocitos con cambios degenerativos asociados y esteatosis focal. Cortesía de los Dres Marcelo Fabián Amante y Gabriel Casas, Servicio de Anatomía Patológica del Hospital Alemán, Buenos Aires.
Figura 3.
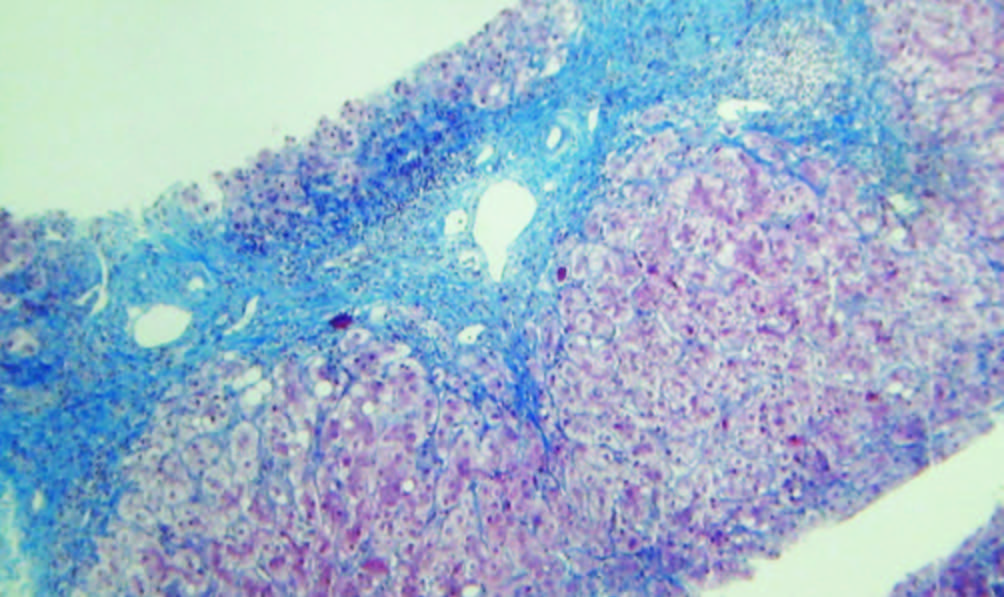
Tricrómico 25 X. La tinción de tricrómico destaca la presencia de puentes fibrosos portoportales. Coexiste infiltrado inflamatorio mononuclear en los tractos fibrosos y espacio porta remanentes. Cortesía de los Dres Marcelo Fabián Amante y Gabriel Casas, Servicio de Anatomía Patológica del Hospital Alemán, Buenos Aires.
Diagnóstico diferencial
En la edad pediátrica debemos descartar además de las hepatitis crónicas virales B o C, la colangitis esclerosante autoinmune, la hepatitis tóxica y la enfermedad de Wilson. La colangitis esclerosante autoinmune comparte el mismo perfil serológico de la HAI tipo 1, pero la primera presenta lesiones de la vía biliar típicas en la colangiografía, lo que permite su identificación.
Hasta el 50% de los pacientes con hepatitis B y C presentan anticuerpos ANA y/o SMA positivos, aunque por lo general en títulos bajos, y hasta un 10% de los pacientes con hepatitis C crónica tienen anticuerpos anti-LKM-1. En estos pacientes, la histología también puede ser similar, pero el grado de inflamación generalmente es menor. La detección de los marcadores virales típicos permite hacer el diagnóstico.
En la enfermedad de Wilson también pueden estar presentes los anticuerpos ANA, y a veces los SMA, en asociación con IgG alta e histología hepática inflamatoria, lo que puede hacer difícil el diagnóstico diferencial con la HAI tipo 1. Los niveles de cobre urinarios, en suero y en el tejido hepático, y la presencia de los anillos de Kayser-Fleischer en el examen oftalmológico con lámpara de hendidura, nos orientan hacia dicho diagnóstico.
En pacientes con presentación aguda, deben además excluirse otras causas virales: hepatitis A y E, citomegalovirus, virus de Epstein-Barr, herpes simple, parvovirus B19 y adenovirus.1-6, 32-35, 46
Tratamiento convencional
El objetivo del tratamiento de la HAI es controlar el proceso inflamatorio del parénquima hepático, con la administración de inmunosupresores. Esta terapia debe iniciarse precozmente, teniendo en cuenta que la rapidez y el grado de respuesta dependen de la severidad de la enfermedad.
El primer esquema terapéutico implementado fue la asociación de prednisona y azatioprina. La prednisona es indicada inicialmente a 2 mg/kg/día (dosis máxima: 40 a 60 mg/d) y la azatioprina a 1,5-2 mg/Kg/día. La dosis de prednisona se reduce gradualmente, siguiendo el descenso de las transaminasas (respuesta terapéutica), con la intención de indicar al paciente la menor dosis capaz de mantener la remisión bioquímica de la enfermedad. La mayoría de los pacientes obtendrán una franca reducción del nivel de transaminasas durante los primeros 2 meses de tratamiento. En algunos casos, la normalización de las mismas será evidente en varios meses más.1-6, 32-36, 46-48
Los efectos adversos relacionados con la administración prolongada de prednisona son: obesidad, hábito cushingoide, estrías cutáneas, hipertricosis, osteopenia/ osteoporosis, diabetes, cataratas, hipertensión arterial, etc. Los vinculados con la administración de azatioprina, aunque infrecuentes, incluyen: colestasis, enfermedad venooclusiva, pancreatitis, toxicidad de médula ósea, etc.
La suspensión del tratamiento inmunosupresor luego de obtenida la remisión y mantenida durante varios años, es un hecho deseable, sin embargo, existen solo reportes anecdóticos. Se menciona una posible discontinuación de la terapia en el 20% de los casos de la HAI tipo 1 pero en ningún niño con la HAI tipo 2.13, 33-37, 46-48
Tratamiento de pacientes no respondedores
Alrededor del 20% de los pacientes con HAI no responden al tratamiento convencional y cerca de un 40% de los respondedores presentan episodios frecuentes de recidivas que requieren un tratamiento alternativo para impedir la progresión de la enfermedad a falla hepática y necesidad de trasplante.
Las alternativas terapéuticas de los casos no respondedores actualmente se basan en drogas inmunosupresoras utilizadas en el post-trasplante para evitar el rechazo celular como el micofenolato mofetil (MMF), inhibidores de la calcineurina y la rapamicina.45
El MMF es un inhibidor de la síntesis de purinas, consiguiendo su efecto inmunosupresor por la disminución en la producción de linfocitos B y T. Se emplea en dosis de 20 mg/kg dos veces al día en aquellos pacientes no respondedores o bien aquellos que no toleran la azatioprina. Los efectos secundarios más comúnmente observados son leucopenia o neutropenia, que a menudo llevan a la necesaria reducción de la dosis o a la suspensión de la medicación. Existen varios reportes de casos sobre el uso del MMF en pacientes refractarios, destacando una serie de 26 pacientes no respondedores al tratamiento clásico, tratados con MMF, seguidos por 5 años, de los cuales el 70% respondió a esta alternativa y manteniéndose con transaminasas normales durante el período de seguimiento.47-49
Si existe intolerancia al MMF o persiste la falta de respuesta, se debe considerar el uso de inhibidores de la calcineurina, tacrolimus o ciclosporina. Éstos deben ser usados con precaución y dosando los niveles en sangre por su potencial toxicidad. Los inhibidores de la calcineurina actúan inhibiendo la expresión del receptor de interleuquina 2 y la expansión de los linfocitos T CD4.
La ciclosporina es un inmunosupresor potente que se ha utilizado efectivamente tanto en niños como en adultos con HAI. En la edad pediátrica se ha demostrado que puede inducir remisión en HAI tipos 1 y 2 al administrarse como tratamiento inicial. Se ha utilizado durante un período de 6 meses, destinado al control del proceso inflamatorio, continuando posteriormente con dosis bajas de prednisona y azatioprina. Se documentó respuesta sostenida en más del 95% de los pacientes con efectos adversos leves y tansitorios.1, 33, 50-53
La rapamicina, un macrólido que inhibe la repuesta a la interleuquina 2 y de esta forma la activación de los linfocitos T y B, también ha sido propuesta para tratamiento de casos de HAI refractaria, aunque la experiencia es limitada.52, 54
Entre las alternativas más recientemente propuestas se encuentra el rituximab. Se trata de un anticuerpo monoclonal quimérico, murino y humano, obtenido por ingeniería genética, que se une específicamente al antígeno CD20, expresado en linfocitos pre-B y B maduros, causando su muerte por apoptosis. El rituximab fue inicialmente aprobado para el tratamiento del linfoma no-Hodgkin y la artritis reumatoidea. Aunque la HAI es considerada una enfermedad autoimmune mediada por células T, las células B han demostrado jugar un rol importante en varias enfermedades autoinmmunes debido a la producción de autoanticuerpos, o como células presentadoras de antígenos, y por la secreción de citoquinas (IL- 2, IF, IL-6). Debido al elevado nivel de IgG y los altos títulos de anticuerpos presentes en HAI, se cree que el rituximab tiene un rol en su tratamiento. Existen algunos trabajos recientemente publicados que demuestran su eficacia en el tratamiento de casos refractarios de HAI con buena tolerancia y un bajo perfil de toxicidad.52, 55-58
El trasplante hepático está indicado en aquellos casos de HAI que se presentan como falla hepática fulminante o bien en los casos en que la enfermedad progresa a insuficiencia hepática terminal a pesar del tratamiento instituido (10-20%), o por abandono del mismo. Luego del trasplante, la tasa de recurrencia de la HAI es cercana al 20%. El diagnóstico de recurrencia se basa en anormalidades bioquímicas, presencia de autoanticuerpos e histología característica. Puede ocurrir varios años post-trasplante; es por ello que se aconseja mantener una dosis de corticoides constante. Debido a la alta tasa de recurrencia y a la intensa inmunosupresión requerida en el post-trasplante en esta situación, es que se deben hacer los mayores esfuerzos para realizar un diagnóstico y tratamiento temprano de la enfermedad con el objeto de evitar, en la mayoría de los casos, la progresión de la misma y la necesidad de trasplante.1-6, 32-37, 59
Agradecimientos. Agradecemos la participación en la revisión bibliográfica de los Doctores Gustavo Boldrini (Argentina), Graciela Caballero (Uruguay), Cristina Galoppo (Argentina) y Violeta Sereno (Uruguay).
Referencias
- Álvarez F. Autoimmune hepatitis and primary sclerosing cholangitis. Clin Liver Dis 2006; 10: 89-107.
- Mieli-Vergani G, Vergani D. Autoimmune paediatric liver disease. World J Gastroenterol 2008; 14: 3360-3367.
- Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, and Vierling JM. AASLD Practice Guidelines. Diagnosis and Management of Autoimmune Hepatitis. Hepatology 2010; 51: 2193-2213.
- Mieli-Vergani G, Heller S, Jara P, Vergani D, Chang MH, Fujisawa T, González-Peralta RP, Kelly D, Mohan N, Shah U, Murray KF. Autoimmune hepatitis. J Pediatr Gastroenterol Nutr 2009; 49: 158-164.
- Mieli-Vergani G, Vergani D. Autoimmune hepatitis. Nat Rev Gastroenterol Hepatol 2011; 8: 320-329.
- Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet 2013; 382: 1433-1444.
- Czaja AJ, Cookson S, Constantini PK, Clare M, Underhill JA, Donaldson PT. Cytokine polymorphisms associated with clinical features and treatment outcome in type 1 autoimmune hepatitis. Gastroenterology 1999; 117: 645-652.
- Cookson S, Constantini PK, Clare M, Underhill JA, Bernal W, Czaja AJ, Donaldson PT. Frequency and nature of cytokine gene polymorphisms in type 1 autoimmune hepatitis. Hepatology 1999; 30: 851-856.
- Kerkar N, Choudhuri K, Ma Y, Mahmoud A, Bogdanos DP, Muratori L, Bianchi F, Williams R Mieli-Vergani G, Vergani D. Cytochrome P4502D6(193-212): a new immunodominant epitope and target of virus/self cross-reactivity in liver kidney microsomal autoantibody type 1-positive liver disease. J Immunol 2003; 170: 1481-1489.
- Liberal R, Longhi MS, Mieli-Vergani G. Pathogenesis of autoimmune hepatitis. Best Pract & Res Clinical Gastroenterol 2011; 25: 653-664.
- Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+) CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol 2004; 41: 31-37.
- Longhi MS, Ma Y, Mitry RR, Bogdanos DP, Heneghan M, Cheeseman P, Mieli-Vergani G, Vergani D. Effect of CD4+ CD25+ regulatory T-cells on CD8 T-cell function in patients with autoimmune hepatitis. J Autoimmun 2005; 25: 63-71.
- Liberal R, Grant CR, Mieli-Vergani G, Vergani D. Autoimmune hepatitis: A comprehensive review. J Autoimmunity 2013; 41: 126-139.
- Longhi MS, Ma Y, Mieli-Vergani G, Vergani D. Aetiopathogenesis of autoimmune hepatitis. J Autoimmunity 2010; 34: 7-14.
- Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A, Baron U, Olek S, Wiegard C, Lohse AW, Weiler- Normann C, Schramm C, Herkel J. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol 2012; 57: 125-132.
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4 + CD25+ regulatory T-cells. Nat Immunol 2003; 4: 330-336.
- Longhi MS, Mitry RR, Samyn M, Scalori A, Hussain MJ, Quaglia A, Mieli-Vergani G, Ma Y, Vergani D. Vigorous activation of monocytes in juvenile autoimmune liver disease escapes the control of regulatory T-cells. Hepatology 2009; 50: 130-142.
- Czaja AJ & Manns MP. Advances in the Diagnosis, Pathogenesis, and Management of Autoimmune Hepatitis. Gastroenterology 2010; 139: 58-72.
- Czaja AJ, Doherty DG, Donaldson PT. Genetic bases of autoimmune hepatitis. Dig Dis Sci 2002; 47: 2139-2150.
- Donaldson PT, Doherty DG, Hayllar KM, McFarlane IG, Johnson PJ, Williams Rl. Susceptibility to autoimmune chronic active hepatitis: human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology 1991; 13: 701-706.
- Yoshizawa K, UmemuraTT, Ota MJ. Genetic background of autoimmune hepatitis in Japan. Gastroenterol 2011; 46: 42-47.
- Goldberg AC, Bittencourt PL, Oliveira LC, Ramasawmy R, Marin ML, Palacios SA, Kalil J, Porta G. Autoimmune Hepatitis in Brazil: An Overview. Scand J Immunol 2007; 66: 208-216.
- Oliveira LC, Porta G, Marin ML, Bittencourt PL, Kalil J, Goldberg AC. Autoimmune hepatitis, HLA and extended haplotypes. Autoimmunity Reviews 2011; 10: 189-193.
- Doherty DG, Underhill JA, Donaldson PT, Manabe K, Mieli-Vergani G, Eddleston AL, Vergani D, Demaine AG, Williams R. Polymorphism in the human complement C4 genes and genetic susceptibility to autoimmune hepatitis. Autoimmunity 1994; 18: 243-249.
- Cançado ELR, Porta G. Autoimmune hepatitis in South América. In: Manns MP, Paumgartner G, Leuschner U. Immunology and liver. Falk Foundation 114. Dordrecht: Kluwer Academic Publishers 2000: 82-92.
- Bittencourt PL, Palácios SA, Cançado EL, Porta G, Carrilho FJ, Laudanna AA, Kalil J, Goldberg AC. Cytotoxic T lymphocyte antigen-4 gene polymorphism do not confer susceptibility to autoimmune hepatitis types 1 and 2 in Brazil. Am J Gastroenterol 2003; 98: 1616-1620.
- Agarwal K, Czaja AJ, Donaldson PT. A functional Fas promoter polymorphism is associated with a severe phenotype in type 1 autoimmune hepatitis characterized by early development of cirrhosis. Tissue Antigens 2007; 69: 227-235.
- Smyk DS, Orfanidou T, Invernizzi P, Bogdanos DP, Lenzi M. Vitamin D in autoimmune liver disease. Clin Res Hepatol Gastroenterol 2013; 37(5): 535-545.
- Lankisch TO, Strassburg CP, Debray D, Manns MP, Jacquemin E. Detection of autoimmune regulator gene mutations in children with type 2 autoimmune hepatitis and extrahepatic immune-mediated diseases. J Pediatr 2005; 146: 839-842.
- Gulati R, Radhakrishnan KR, Hupertz V, Wyllie R, Alkhouri N, Worley S, Feldstein AE. Health-related quality of life in children with autoimmune liver disease. J Pediatr Gastroenterol Nutr 2013; 57: 444-450.
- Liberal R, Mieli-Vergani G, Vergani D. Clinical significance of autoantibodies in autoimmune hepatitis. J Autoimmun 2013; 46: 17-24.
- Lohse AW, Mieli-Vergani G. Autoimmune hepatitis. J Hepatol 2011; 55: 171-182.
- Cuarterolo M, Ciocca M, Alvarez F. Hepatitis autoinmune en niños. Perpectivas actuales. Arch Argent Pediatr 2014; 112: 169-175.
- Mieli-Vergani G, Vergani D. Autoimmune hepatitis in children. Clin Liver Dis 2002; 6: 335-346.
- Galicia Poblet G, López-Manzanares MJ. Protocolos Diagnóstico- Terapéuticos de Gastroenterología, Hepatología y Nutrición Pediátrica 2010. SEGHNP-AEP. Hepatitis Autoinmune: 211-220.
- Gleeson D, Heneghan MA. British Society of Gastroenterology (BSG) guidelines for management of autoimmune hepatitis. Gut 2011; 60: 1611-1619.
- Mieli-Vergani G, and Vergani D. Autoimmune hepatitis in children: what is different from adult AIH? Seminars in liver disease 2009; 29: 297-306.
- Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001; 344: 495-500.
- Scheuer PJ. Liver biopsy matters in chronic hepatitis: bigger is better. Hepatology 2003; 38: 1357-1358.
- Suzuki A, Brunt EM, Kleiner DE, Miguel R, Smyrk TC, Andrade RJ, Lucena MI, Castiella A, Lindor K, Björnsson E. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury. Hepatology 2011; 54: 931-939.
- Kumari N, Poddar U, Srivastav A, Kathuria R, Krishnani N, Yachha SK. Significance of histopathological features in differentiating autoimmune liver disease from nonautoimmune chronic liver disease in children. European Journal of Gastroenterology & Hepatology 2013; 25: 333-337.
- Stravitz RT, Lefkowitch JH, Fontana RJ, Gershwin ME, Leung PS, Sterling RK, Manns MP, Norman GL, Lee WM; Acute Liver Failure Study Group. Autoimmune Acute Liver Failure: Proposed Clinical and Histological Criteria. Hepatology 2011; 53: 517-526.
- Rojas CP, Bodicharla R, Campuzano-Zuluaga G, Hernandez L, Rodriguez MM. Autoimmune hepatitis and primary sclerosing colangitis in children and adolescents. Fetal and Pediatric Pathology 2014: 33: 202-209.
- Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, Desmet V, Korb G MacSween RN. Histological grading and staging of chronic hepatitis. J Hepatol 1995; 22: 696-699.
- Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. Hepatology 2004; 40: 646-652.
- Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, Donaldson PT, Eddleston AL, Fainboim L, Heathcote J, Homberg JC, Hoofnagle JH, Kakumu S, Krawitt EL, Mackay IR, MacSween RN, Maddrey WC, Manns MP, McFarlane IG, Meyer zum Büschenfelde KH, Zeniya M. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999; 31: 929-938.
- Vergani D, Mieli-Vergani G. Pharmacological management of autoimmune hepatitis. Expert Opin Pharmacother 2011; 12: 607-613.
- Della Corte C, Sartorelli MR, Sindoni CD, Girolami E, Giovannelli L, Comparcola D, Nobili V. Autoimmune hepatitis in children: an overview of the disease focusing on current therapies. Eur J Gastroenterol Hepatol 2012; 24: 739-746.
- Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli-Vergani G. Mycophenolate mofetil as rescue treatment for autoimmune liver disease in children: a 5-year follow-up. J Hepatol 2009; 51: 156-160.
- Álvarez F, Ciocca M, Cañero-Velasco C, Ramonet M, de Dávila MT, Cuarterolo M, González T, Jara-Vega P, Camarena C, Brochu P, Drut R, Álvarez E. Short-term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol 1999; 30: 222-227.
- Casswall TH, Beijer E, Németh A. Tacrolimus without or with the addition of conventional immunosuppressive treatment in juvenile autoimmune hepatitis. Acta Paediatr 2012; 101: 993-999.
- Czaja AJ. Nonstandard drugs and feasible new interventions for autoimmune hepatitis: part I. Review. Inflamm Allergy Drug Targets 2012; 11: 337-350.
- Tannous MM, Cheng J, Muniyappa K, Farooq I, Bharara A, Kappus M, Luketic V, Stravtiz RT, Fuchs M, Puri P, Sanyal A, Sterling R. Use of tacrolimus in the treatment of autoimmune hepatitis: a single centre experience. Aliment Pharmacol Ther 2011; 34: 405-407.
- Chatrath H, Allen L, Boyer TD. Use of sirolimus in the treatment of refractory autoimmune hepatitis. Am J Med 2014; 127: 1128-1131.
- Mieli-Vergani G, Vergani D. Paediatric autoimmune liver disease. Arch Dis Child 2013; 98: 1012-1017.
- D’Agostino D, Costaguta A, Álvarez F. Successful treatment of refractory autoimmune hepatitis with rituximab. Pediatrics 2013; 132: e526-e530.
- Burak KW, Swain MG, Santodomingo-Garzon T, Lee SS, Urbanski SJ, Aspinall AI, Coffin CS, Myers RP. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can J Gastroenterol 2013; 27: 273-280.
- Al-Busafi SA, Michel RP, Deschenes M. Rituximab for refractory autoimmune hepatitis: a case report. Arab J Gastroenterol 2013; 14: 135-138.
- Zahr Eldeen F, Mabrouk Mourad M, Liossis C, Bramhall SR. Liver retransplant for primary disease recurrence. Exp Clin Transplant 2014; 12: 175-183.
Correspondencia: Mirta Ciocca
Hospital Alemán, Av Pueyrredón 1640. Ciudad Autónoma de Buenos
Aires, Argentina
Correo electrónico: mciocca@intramed.net
Acta Gastroenterol Latinoam 2016;46(3): 237-245
